
Highly scalable production of uniformly-coated superparamagnetic nanoparticles for triggered drug release from alginate hydrogels†
Abstract
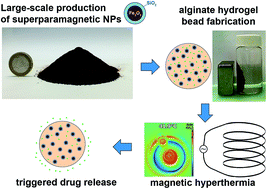
Intelligent, on-demand drug administration systems with controlled release kinetics may revolutionize the way diseases are treated. Typically, the release of the therapeutic payload from these systems is activated by stimuli-responsive nanofillers. However, limitations regarding large-scale nanomaterial production and poor reproducibility keep such systems in the labs and away from clinics. Here, we demonstrate the highly scalable and reproducible synthesis of uniform superparamagnetic Fe2O3 nanoparticles coated with a nanothin layer of amorphous SiO2, and evaluate their suitability as stimuli-responsive nanofillers in a drug-loaded biopolymer alginate matrix. The superior colloidal stability of the SiO2-coated Fe2O3 nanoparticles over their uncoated counterparts and their dispersibility in aqueous suspensions facilitates their incorporation in alginate hydrogel microbeads. We examine the hyperthermia performance of such multiscale particle structures in the presence of alternating magnetic fields and compare the release of dextran (a model biomolecule) in the presence and absence of external stimuli. The enhanced triggered release of dextran in the presence of magnetic fields further highlights the potential of such superparamagnetic SiO2-coated Fe2O3 nanoparticles as a functional transducer in such systems.
 Please wait while we load your content...
Please wait while we load your content...

