
Enhanced anode performance of manganese oxides with petal-like microsphere structures by optimizing the sintering conditions†
Abstract
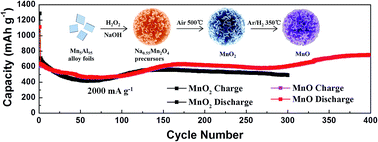
Herein, the rational design and synthesis of manganese oxides (MnO2 and MnO) have been achieved and both of them show petal-like microsphere structures. As anodes for LIBs, MnO exhibits a higher capacity of 751.4 mA h g−1 after 400 cycles (492.7 mA h g−1 for MnO2 after 300 cycles) at 2000 mA g−1.
 Please wait while we load your content...
Please wait while we load your content...

