
Nonclassical from-shell-to-core growth of hierarchically organized SAPO-11 with enhanced catalytic performance in hydroisomerization of n-heptane†
Abstract
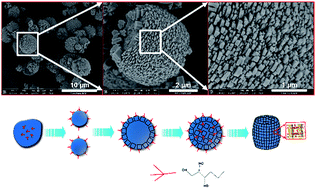
Integrating hierarchical porosity over microporous zeotype materials is an effective way to promote their mass transfer properties and catalytic performances. A combined synthetic strategy using small molecular growth inhibitor 1,2,3-hexanetriol and tumbling crystallization condition to generate hierarchically organized SAPO-11 is herein presented. The addition of 1,2,3-hexanetriol in the synthetic gel of SAPO-11 under agitating conditions significantly altered its crystallization behaviour, resulting in the formation of a hierarchically organized architecture. An underlying nonclassical from-shell-to-core crystallization has been disclosed by time-dependent observation of the formation process. The hierarchically self-organized structure has been characterized by a suite of characterization techniques, such as XRD, N2 physisorption, SEM, TEM, mercury intrusion measurements, 27Al, 29Si, 31P MAS NMR and pyridine adsorption IR (Py-IR). The structure featuring barrel-shaped architecture is comprised of aligned 300–400 nm primary building blocks with voids in between, constructing an auxiliary macro-/meso-pore system open to external surfaces. The catalytic performance of Pt supported on hierarchical SAPO-11 in n-heptane hydroisomerization has been assessed, showing that both catalytic activity and isomer yield have been increased with respect to a conventional sample. As the acidity for the hierarchical SAPO-11 is comparable to the conventional sample, the enhancement in catalytic performance is attributed to the small primary crystal size and macro-/meso-pore-connectivity, that are important for mass transfer.
 Please wait while we load your content...
Please wait while we load your content...

