
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Invasin-functionalized liposome nanocarriers improve the intracellular delivery of anti-infective drugs†
Sara
Menina
ab,
Hagar Ibrahim
Labouta‡
a,
Rebecca
Geyer
c,
Tanja
Krause
c,
Sarah
Gordon
a,
Petra
Dersch
c and
Claus-Michael
Lehr
*ab
aDepartment of Drug Delivery, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Center for Infection Research (HZI), Saarbruecken, Germany. E-mail: Claus-Michael.Lehr@helmholtz-hzi.de
bDepartment of Pharmacy, Saarland University, Saarbruecken, Germany
cDepartment of Molecular Infection Biology, Helmholtz Center for Infection Research (HZI), Braunschweig, Germany
First published on 20th April 2016
Abstract
Intracellular infections caused by invasive pathogens continue to prove difficult to combat, due in part to the commonly poor membrane permeability of anti-infective drugs. The aim of this study was to improve the intracellular delivery of one such poorly permeable (but broad-spectrum) anti-infective, gentamicin. Gentamicin was encapsulated into liposomal nanocarriers which were then surface functionalized with InvA497, a bacteria-derived invasion protein. Treatment of HEp-2 cells infected with the enteroinvasive bacteria Yersinia pseudotuberculosis or Salmonella enterica with gentamicin-containing, InvA497-functionalized liposomes resulted in a significantly greater reduction in infection load than treatment with non-functionalized liposomes, indicating that such a bacteriomimetic nanocarrier was not only able to promote successful cellular uptake of gentamicin but was also able to mediate anti-infective drug delivery to both cell cytoplasm and intracellular compartments. The developed InvA497-functionalized liposomal nanocarrier therefore holds great promise as a strategy for improving the therapy of intracellular infections.
1. Introduction
As clearly demonstrated by the ever-increasing incidence of ‘hard-to-treat’ infections,1 the ability of pathogens to evade immune detection and resist the action of anti-infective agents is continually evolving. Effective survival strategies have been adopted by numerous pathogens, including bacteria such as Mycobacterium tuberculosis, Staphylococcus aureus and Salmonella enterica. These bacteria invade mammalian cells and take refuge either in the cytosol, or in other subcellular shelters such as membrane-bound vacuoles.2 This invasion strategy not only provides concealment from the host immune system but also confers protection against anti-infective agents. This is based on the fact that a considerable number of anti-infective drugs exhibit either a poor permeability into mammalian cells due to their high degree of hydrophilicity, or a poor level of intracellular retention.3 Their administration may therefore result in sub-therapeutic intracellular concentrations of anti-infectives, providing a driving stimulus for the development of bacterial resistance. Furthermore, the use of high-dose anti-infective therapies in an effort to increase intracellular drug concentrations may result in adverse or even toxic outcomes.4 The effective intracellular delivery of anti-infective agents with poor permeability characteristics therefore clearly constitutes a considerable and ongoing challenge for the treatment of infectious diseases.Encapsulation of anti-infective drugs within nanoscale delivery systems has been shown to provide a means to enhance the permeation of these compounds into mammalian cells, while also allowing for control over drug release kinetics and minimization of drug-associated side effects.5 Liposomes in particular, as biocompatible, biodegradable delivery systems, are reported as being the most commonly utilized nanocarriers for the intracellular delivery of anti-infectives to date.6,7 A further enhancement of intracellular access may be achieved by surface functionalization of nanocarriers such as liposomes with specific cell-targeting moieties.8 However, despite the considerable potential for augmentation of nanocarrier-mediated intracellular delivery through the use of such moieties, as often demonstrated for example in tumor cell targeting, surface modification of anti-infective-loaded liposomes with invasive molecules is not common practice.4 As such, this remains a strategy to be fully exploited.
In this respect, the use of bacteria-derived proteins known to mediate cellular adhesion and invasion is of considerable interest and potential, as functionalization of nanocarriers with such proteins would allow for exploitation of the natural mechanisms utilized by invasive bacteria to induce their own uptake and establish intracellular infection.9 One of the most well characterized bacteria-derived invasion factors to date is invasin, an outer membrane protein expressed by members of the Yersinia spp. (e.g. Y. pseudotuberculosis and Y. enterocolitica) which mediates bacterial entry into mammalian cells through interaction with cellular β1 integrin receptors.10–12 Binding of invasin to cell surface integrin receptors and subsequent receptor activation leads to a re-organization of the host cell cytoskeleton and the formation of cytoplasmic protrusions, ultimately resulting in engulfment of the invasin-expressing bacterium and intracellular encapsulation within a membrane-bound vacuole.13,14
Previously, the potential of invasin-functionalized particulate nanocarriers to affect intracellular delivery of a model vaccine antigen has been demonstrated;15,16 surface functionalization of latex or polystyrene nano- or micro-particles with invasin has also been shown to facilitate an enhanced uptake of particles into both phagocytic and non-phagocytic mammalian cells.8,17–19 A recent study has presented mechanistic insights into the interaction of invasin-functionalized liposomes with β1 integrin receptor-expressing epithelial cells, and provided preliminary indications of the efficacy of such a system in facilitating intracellular anti-infective delivery.20 Yet, in-depth studies focused on fabrication of anti-infective-loaded, invasin-functionalized nanocarriers together with detailed assessment of antimicrobial efficacy against intracellular pathogens remain to be performed. The current work therefore aimed to constitute such a study.
To this end, liposomes as highly biocompatible nanocarriers were first loaded with gentamicin, a poorly permeable anti-infective with a broad spectrum of antibacterial action, using different preparation methods in order to achieve optimal drug loading and system stability. Liposomes were then surface functionalized with the C-terminal region of invasin (InvA497), and further characterized. The ability of gentamicin-loaded, InvA497-functionalized liposomes to deliver gentamicin to the interior of cells and subcellular compartments was then determined, by conducting efficacy studies employing the human epithelial cell line HEp-2 infected with the common enteropathogenic bacteria S. enterica and Y. pseudotuberculosis.
2. Experimental
2.1. Liposome preparation and drug loading
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.6:3, together with 10 μg ml−1 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (Liss Rhod PE, Avanti Polar Lipids Inc., Alabaster, USA), were dissolved in a round-bottomed flask in a mixture of chloroform and methanol (2:1 w/w). The solvent was evaporated to dryness under reduced pressure at 70 °C, 200 mbar and 145 rpm. The formed thin lipid film was then hydrated by the addition of 5 ml of either phosphate buffered saline (PBS, pH 7.4) or gentamicin solution (10 mg ml−1 in PBS, Sigma Aldrich, Steinheim, Germany) for 1 h with continual rotation, in order to form unloaded or gentamicin-loaded liposomes respectively. Liposomes were then extruded 10 times through 200 nm pore size polycarbonate membranes (Sartorius, Goettingen, Germany) using a 50 ml extruder (LiposoFast L-50 Avestin, Mannheim, Germany) to facilitate size reduction. The liposomal dispersion was then diluted 1:10 with PBS and stored at 4 °C. Liposomal purification was carried out prior to any further investigations (see ‘Liposome purification’ below).
:3 in PBS and stirred at 520 rpm and 30 °C in order to form a water-in-oil-in-water emulsion. Stirring was continued until the organic solvent was completely evaporated (60 min). The liposomal dispersion was finally extruded, diluted, stored and later purified as described for the LFH method.
000g for 45 min at 4 °C. Pelleted liposomes were re-suspended in carbonate buffer (pH 10.2) containing 10 mg ml−1 of gentamicin, and incubated at 37 °C for 1 h with intermittent vortex mixing in order to facilitate gentamicin loading. Liposome extrusion, dilution, storage and later purification were carried out as described.
:0.6:3, together with 10 μg ml−1 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (Liss Rhod PE, Avanti Polar Lipids Inc., Alabaster, USA), were dissolved in a round-bottomed flask in a mixture of chloroform and methanol (2:1 w/w). The solvent was evaporated to dryness under reduced pressure at 70 °C, 200 mbar and 145 rpm. The formed thin lipid film was then hydrated by the addition of 5 ml of either phosphate buffered saline (PBS, pH 7.4) or gentamicin solution (10 mg ml−1 in PBS, Sigma Aldrich, Steinheim, Germany) for 1 h with continual rotation, in order to form unloaded or gentamicin-loaded liposomes respectively. Liposomes were then extruded 10 times through 200 nm pore size polycarbonate membranes (Sartorius, Goettingen, Germany) using a 50 ml extruder (LiposoFast L-50 Avestin, Mannheim, Germany) to facilitate size reduction. The liposomal dispersion was then diluted 1:10 with PBS and stored at 4 °C. Liposomal purification was carried out prior to any further investigations (see ‘Liposome purification’ below).
:3 in PBS and stirred at 520 rpm and 30 °C in order to form a water-in-oil-in-water emulsion. Stirring was continued until the organic solvent was completely evaporated (60 min). The liposomal dispersion was finally extruded, diluted, stored and later purified as described for the LFH method.
000g for 45 min at 4 °C. Pelleted liposomes were re-suspended in carbonate buffer (pH 10.2) containing 10 mg ml−1 of gentamicin, and incubated at 37 °C for 1 h with intermittent vortex mixing in order to facilitate gentamicin loading. Liposome extrusion, dilution, storage and later purification were carried out as described.
2.2. Surface functionalization of liposomes with InvA497
Covalent coupling of InvA497 (produced as described in the ESI†) to LFH liposomal surfaces was performed as described previously.20,25 Briefly, 2 ml of LFH liposomal suspension was incubated for a minimum of 3 h with a crosslinking reagent solution, consisting of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, 48 mM, Sigma Aldrich, Steinheim, Germany) and N-hydroxysuccinimide (NHS, 19 mM, Carbolution Chemicals, Saarbruecken, Germany) in an ice bath with gentle mixing. Liposomes were then washed (described below) to remove excess crosslinking reagent. A volume of 300 μl of InvA497 (1 mg ml−1 in PBS) was then added, followed by incubation in an ice bath overnight with gentle mixing. Further washing of liposomes was then carried out to remove non-conjugated invasin.2.3. Liposome purification
Purification of liposomes was carried out by centrifugal ultrafiltration using Centrisart® tubes (Sartorius, Goettingen, Germany) equipped with a 300000 molecular weight cut off (MWCO) membrane. Briefly, the liposomal suspension was placed into a Centrisart® tube followed by the filtration membrane, and centrifuged at 3720g and 4 °C for 30 min. The ultra-filtrate was then removed and the liposomes were re-suspended in fresh PBS buffer. In all cases, 3 purification cycles were performed to ensure the complete removal of any residual non-liposomal material.
2.4. Physical characterization
The size distribution and zeta potential of liposomes were measured by photon correlation spectroscopy and electrophoretic mobility respectively, using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK). In all cases liposomes were dispersed in PBS pH 7.4 prior to measurement, and measurements were conducted at 25 °C.2.5. Gentamicin encapsulation efficiency and loading capacity
A fluorometric method based on the use of o-phthaldialdehyde reagent (OPA, Sigma Aldrich, Steinheim, Germany) was used to quantify gentamicin encapsulated within liposomes. Gentamicin was first extracted from liposomal nanocarriers and processed for analysis by treatment with OPA as previously described.26 The fluorescence of processed samples was then measured on a fluorimeter (Tecan Infinite M200, Maennedorf, Switzerland) at an excitation wavelength of 344 nm and an emission wavelength of 450 nm. The amount of gentamicin contained within samples was determined with reference to standards and used to calculate nanocarrier encapsulation efficiency;27 liposomal gentamicin content was also employed together with the total amount of liposomal lipid content (determined as described in the ESI†) in order to calculate loading capacity.282.6. Quantification of InvA497 functionalization
The amount of InvA497 coupled to liposomal surfaces was quantified using a bicinchoninic acid protein assay (BCA) kit, in accordance with the instructions of the manufacturer (Sigma Aldrich, Steinheim, Germany). The amount of surface-coupled InvA497 was quantified by measurement of UV absorbance at 562 nm in reference to created standard curves, and expressed as a functionalization efficiency (%) relative to the initial amount of InvA497 added.29 A loading ratio (coupled InvA497:liposomal lipid content, % w/w) was also calculated.
2.7. Gentamicin release kinetics
The in vitro release behavior of gentamicin from liposomes was investigated using a dialysis-based method.30 Briefly, following verification with gentamicin solutions that drug transport was not a limiting factor in the employed dialysis setup, samples of gentamicin-containing liposomes either with or without InvA497 surface functionalization (each containing 0.325 mg gentamicin) were added to 10000 MWCO dialysis bags (Spectrum Laboratories Inc., California, USA). Bags were sealed and suspended in 30 ml of PBS (pH 7.4) as release medium, at 37 °C with continuous shaking. At predetermined time intervals, 1 ml of PBS was withdrawn for analysis of gentamicin content and replaced with an equal volume of fresh PBS. Cumulative release of gentamicin was then calculated by quantification of gentamicin in removed PBS aliquots, using the OPA fluorometric method described above.
2.8. Cell culture
Cells of the human larynx carcinoma-derived HEp-2 cell line were cultured in a 75 cm2 flask using Roswell Park Memorial Institute (RPMI 1640) medium (Gibco, Paisley, UK), supplemented with 7.5% newborn calf serum (NCS). Cells were incubated in a humidified incubator (Heraeus CO2 Thermo Scientific Incubator) at 37 °C and 5% CO2. Medium was changed every two days and cells were split upon confluency.2.9. Determination of anti-infective efficacy
2.10. Statistical analysis
Where appropriate, data are shown as mean ± standard error of the mean (SEM) from at least three independent experiments. One way ANOVA followed by post hoc analysis or Student's t-test were performed where relevant to determine the statistical significance of any observed differences. Differences were considered to be significant (*) at a p-value <0.05.3. Results
3.1. Physical characteristics
In order to investigate the optimal method for production of gentamicin-loaded liposomes, nanocarriers were prepared using three different production procedures. Passive loading of gentamicin by liposomal preparation via the LFH method resulted in liposomes with a mean initial size of approximately 202 nm (Fig. 1A and S1A†). Liposomes prepared with the MCV method, also utilizing passive drug loading, had a size of approximately 137 nm, while a slightly larger mean liposomal size (approximately 244 nm) was seen following active loading of gentamicin using the ASL method (Fig. 1A). The polydispersity index (PDI) of all three preparations was below 0.2 (Fig. 1B) and a highly negative surface charge was also noted initially in all cases (Fig. 1C). The three preparations were then stored at 4 °C and evaluated over time in order to provide an assessment of colloidal stability; no appreciable change in size, PDI and zeta potential was noted for any of the nanocarriers over a period of 21 days (Fig. 1A–C).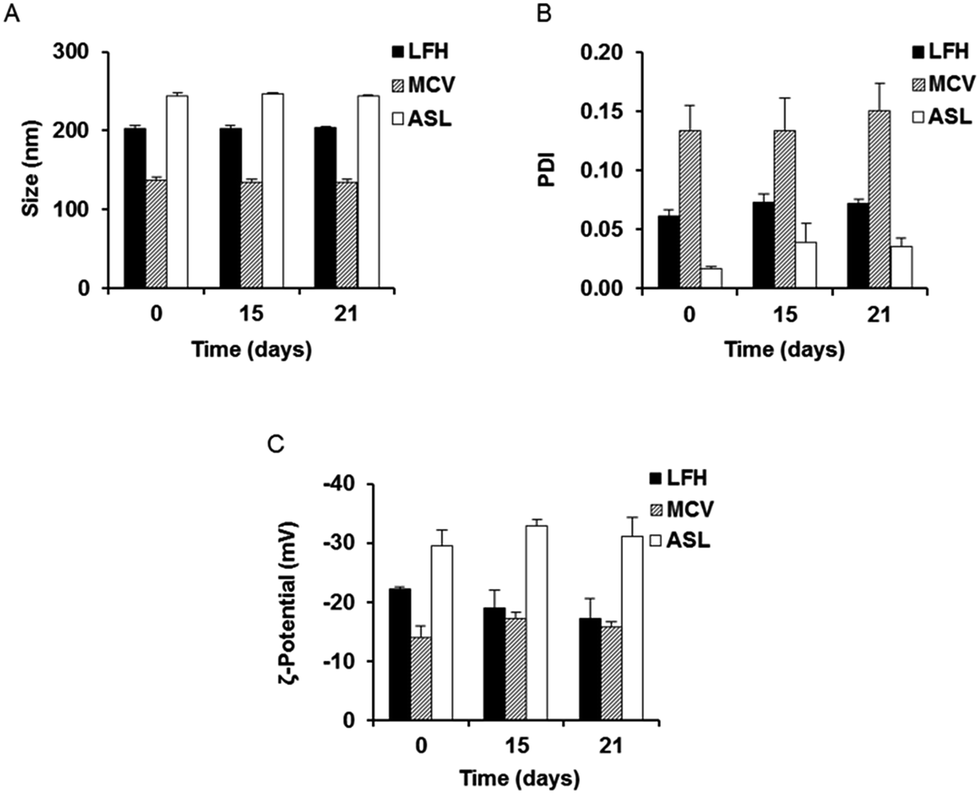 | ||
| Fig. 1 (A) Size, (B) polydispersity index (PDI) and (C) zeta potential of liposomes produced by lipid film hydration (LFH), microencapsulation vesicle (MCV) and ammonium sulfate liposome (ASL) methods were measured at day 0, day 15, and day 21. Values represent the mean ± SEM values from three independent preparations. | ||
3.2. Encapsulation efficiency and loading capacity
The extent of liposome drug encapsulation and loading was also determined initially and monitored over time. LFH liposomes showed the highest initial encapsulation efficiency of approximately 43%, which was significantly higher than that of the ASL liposomes, while the encapsulation efficiency of MCV was approximately 33% (Fig. 2A). In contrast to the encapsulation efficiency, the initial loading capacity of ASL liposomes appeared to be the highest of the three preparations at approximately 34% – however, this was not a statistically significant result. No significant difference was also noted between the initial loading capacity of LFH liposomes (30%) and MCV liposomes (25%) (Fig. 2B). After 15 days, both the encapsulation efficiency and loading capacity of LFH liposomes had decreased, to approximately 15% and 11% respectively, while only 5% encapsulation efficiency and 2% loading capacity were found for MCV liposomes at the same time point. Surprisingly, the ASL preparation was not found to contain any gentamicin after 15 days. At day 21, only the LFH liposomes were seen to retain gentamicin, with no drug detected in MCV and ASL preparations.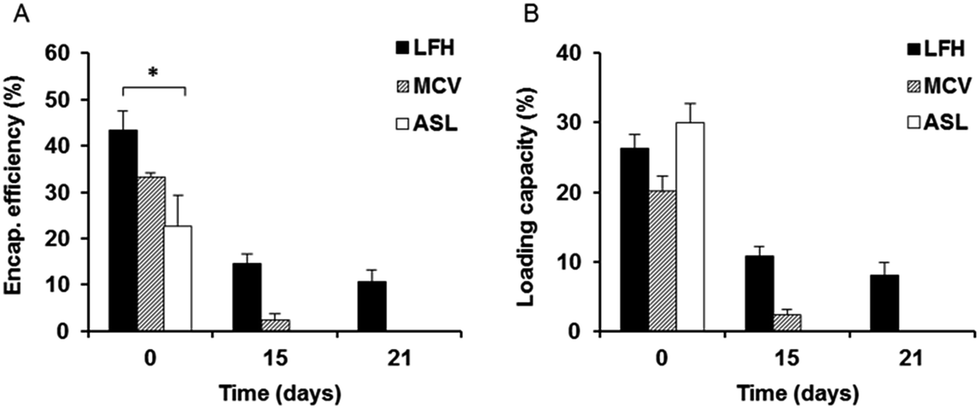 | ||
| Fig. 2 Stability of liposome encapsulation efficiency (A) and loading capacity (B) at day 0, day 15 and day 21. Values represent the mean ± SEM values from three independent preparations. Significance was defined as * = p value <0.05. | ||
As the LFH liposomes showed the highest encapsulation efficiency and the greatest ability to sustain gentamicin encapsulation over time, this preparation was utilized for further experiments.
3.3. Characterization of InvA497-functionalized liposomes
Gentamicin-loaded LFH liposomes functionalized with InvA497 were subjected to initial characterization followed by a short-term stability study. Results showed homogeneously sized liposomes of approximately 198 nm in diameter, with a zeta potential of approximately −23 mV (Fig. 3 and S1B†). These characteristics were similar to those of non-functionalized liposomes, meaning that the functionalization process did not have an appreciable effect on liposome colloidal properties. After 7 days of storage, the size, PDI and zeta potential of InvA497-functionalized liposomes showed no significant changes (Fig. 3A–C). The efficiency of carbodiimide-mediated crosslinking of InvA497 to liposomal surfaces was measured via a BCA assay, with results confirmed by SDS-PAGE (data not shown). An initial InvA497 functionalization efficiency of approximately 60% was found (Fig. 3D), while the ratio of coupled InvA497 to total liposomal lipid content (% w/w) was approximately 17%. InvA497 functionalization of liposomes was seen to result in a considerable reduction in gentamicin encapsulation efficiency, from approximately 40% for non-functionalized liposomes (Fig. 2A, LFH) to approximately 15% for functionalized liposomes (Fig. 3D). The encapsulation efficiency at day 2 decreased to 7%, and further to 5% after 7 days. The functionalization efficiency was also seen to drop upon storage (Fig. 3D).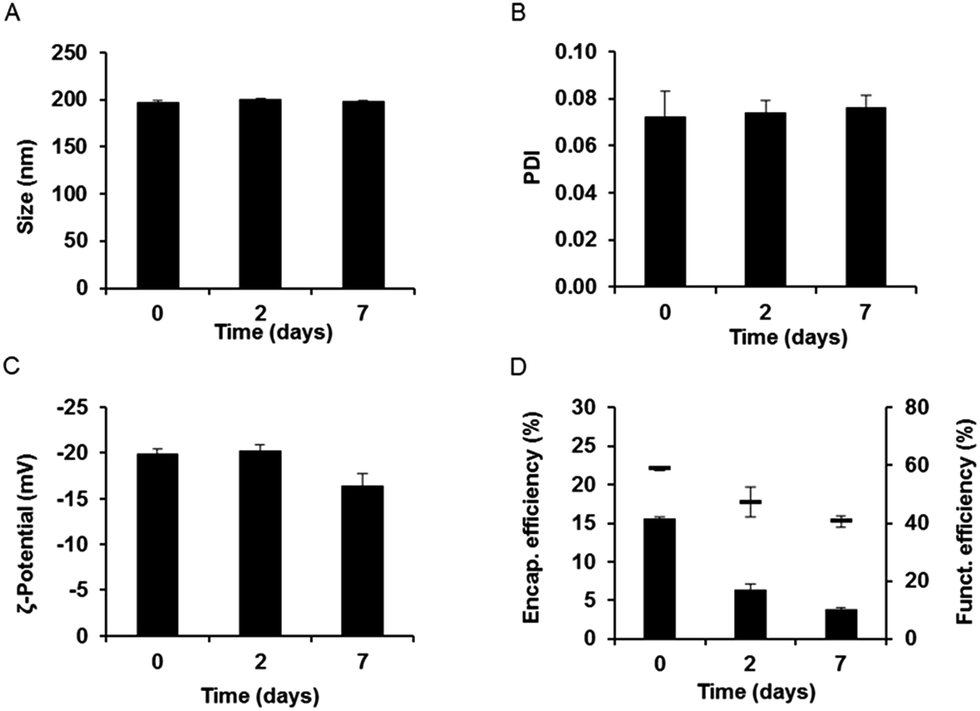 | ||
| Fig. 3 Colloidal stability after functionalization was evaluated by measuring size (A), polydispersity index (PDI) (B) and zeta potential (C) at days 0, 2 and 7. Chemical stability was evaluated by measurement of the stability of gentamicin encapsulation efficiency (bars) and InvA497 functionalization efficiency (lines) (D). Results represent mean ± SEM of three independent preparations. | ||
3.4. Gentamicin release kinetics
The kinetics of gentamicin release from non-functionalized gentamicin-loaded liposomes (GL) and InvA497-functionalized gentamicin loaded liposomes (I-GL) dispersed in PBS at physiological temperature was investigated (Fig. 4). An initial burst release of gentamicin was seen upon dispersion in PBS. As a result, approximately 65% of the originally entrapped gentamicin was released from both GL and I-GL nanocarriers within a period of 3 h. No significant difference in release from GL and I-GL nanocarriers was however noted, indicating that the functionalization procedure had no effect on gentamicin release kinetics.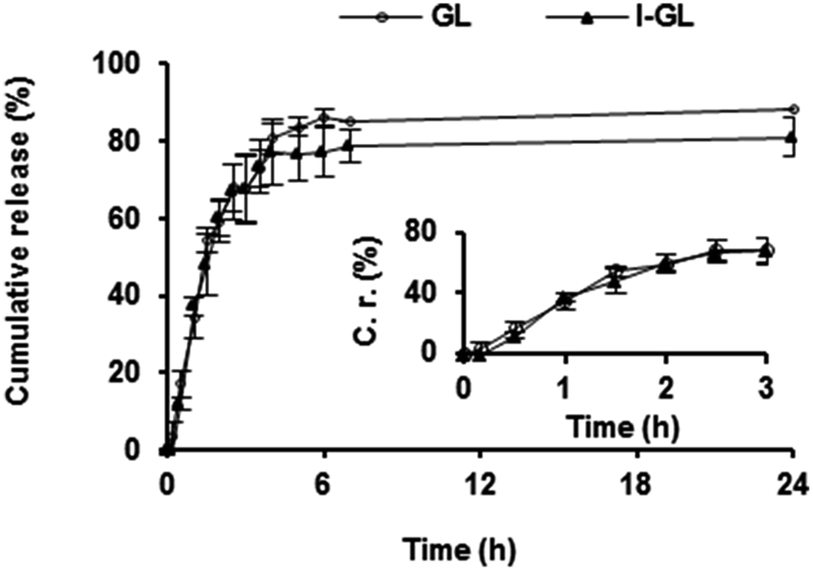 | ||
| Fig. 4 In vitro release profile of gentamicin from liposomes functionalized with InvA497 (I-GL) or without surface functionalization (GL), over a 24 h time period. The inset graph shows release over the first 3 h. Data represent the mean of 3 independent replicate experiments, while error bars are representative of SEM values. | ||
3.5. Determination of anti-infective efficacy
:HEp-2 cells) of 10 with 1 h of infection time and 1 h of gentamicin solution treatment for extracellular bacteria killing (EBK) resulted in the highest cell invasion load for S. enterica (Fig. 5A). For Y. pseudotuberculosis, employment of an MOI of 25 together with 1 h infection time and 1 h of EBK resulted in a similar invasion percentage to that of an MOI of 10, with 1 h infection time and 1 h EBK (Fig. 5B). Thus, for the conduction of anti-infective efficacy studies, invasion conditions of an MOI of 10, 1 h infection time and 1 h of EBK were chosen for S. enterica, while an MOI equal to 25 (in order to maximize the number of bacteria) with 1 h infection time and 1 h of EBK was chosen for optimal Y. pseudotuberculosis invasion.
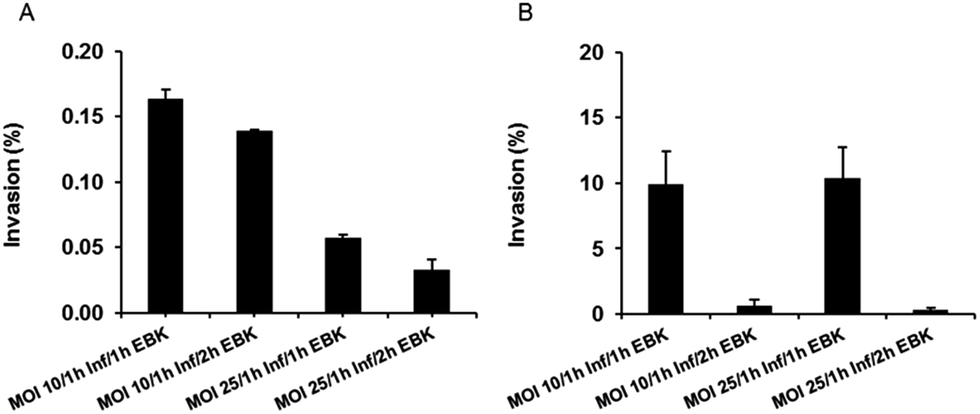 | ||
| Fig. 5 Percentage of recovered intracellular S. enterica (A) and Y. pseudotuberculosis (B) after infection of HEp-2 cells at MOIs of 10 and 25 respectively, followed in all cases by 1 h infection time (Inf), and an extracellular bacteria killing (EBK) of either 1 or 2 h. Data represent the mean ± SEM of three independent preparations. | ||
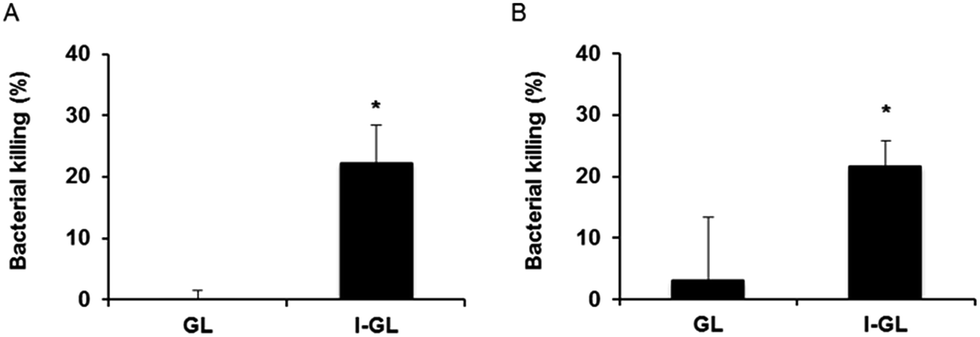 | ||
| Fig. 6 HEp-2 cells infected with S. enterica (A) or Y. pseudotuberculosis (B) treated with gentamicin liposomes (GL) and InvA497-functionalized gentamicin liposomes (I-GL). Data for bacterial killing are normalized to values of untreated cells. Values represent the mean ± SEM from three independent experiments. Significance was defined as * = p value <0.05. | ||
4. Discussion
4.1. Physico-chemical characteristics of liposomal nanocarriers
In the current work, liposomes loaded with the poorly permeable anti-infective gentamicin were formulated in order to facilitate drug penetration into mammalian epithelial cells, allowing for targeting of invasive, intracellular bacteria. Three different preparation methods were initially utilized for the production of gentamicin-loaded liposomes: two methods in which gentamicin was passively loaded during vesicle formation (LFH and MCV), and a third method in which drug was actively incorporated into liposomes post-production (ASL). Liposomes prepared by each method were monitored with respect to both their colloidal and chemical characteristics, in order to determine which nanocarrier system exhibited the most favorable properties both initially and over time. This comparison of preparation approaches was deemed to be an important first step as hydrophilic drugs such as gentamicin are notoriously difficult to retain within nanocarriers, having the tendency to escape rapidly to the external aqueous phase during and/or following the particle preparation process.31–33 Liposomes prepared via the LFH technique were selected for further use following such investigation, as in addition to favorable colloidal properties (exhibited in fact by all produced liposomal nanocarriers), LFH liposomes showed the highest initial encapsulation efficiency and the greatest capacity to sustain incorporation of gentamicin over time upon storage – important points of distinction considering subsequent functionalization of liposomes with InvA497 would necessitate several incubation periods, in addition to washing cycles. The obtained results are in agreement with published literature, where it has been shown that passive loading of liposomes with gentamicin is preferable to active loading, due to the resulting higher amounts of entrapped drug and drug-to-lipid ratio.34 Additional insight is however provided by the current work, which, by conducting a direct comparison of LFH and MCV methodologies, has shown that the specific selection of passive loading method is also important in order to maximize liposomal drug encapsulation and loading.Liposomes prepared by the LFH method were then successfully surface-functionalized with the aforementioned InvA497, a C-terminal fragment of the Y. pseudotuberculosis outer membrane protein invasin.10 The size, PDI and zeta potential of InvA497-functionalized liposomes were similar to that of non-functionalized liposomes; however, the functionalization procedure was seen to have a noticeable effect on the gentamicin encapsulation efficiency, which dropped from approximately 40% to approximately 15%. This decrease, likely due to the mentioned multiple washing steps required and employed during the functionalization procedure, is nevertheless still higher than what has been found previously in some cases using the LFH method,34–36 and was not deemed as being prohibitive to the conduction of further studies. The functionalized liposomal nanocarriers showed a colloidal stability over one week, while the amount of the encapsulated drug was seen to decrease over this period. A focus is therefore currently being placed on optimization of the functionalization protocol; freshly prepared liposomes were meanwhile utilized for all further studies in the currently presented work in order to have maximal drug incorporation.
In addition to the capacity of functionalized and non-functionalized liposomes to retain encapsulated gentamicin on storage, the kinetics of gentamicin release from such nanocarriers in more physiologically-relevant conditions was also of interest. The in vitro release kinetics of gentamicin from liposomes either functionalized or not functionalized with invasin – I-GL and GL respectively – were therefore investigated. Approximately 65% of the total liposomal gentamicin was seen to be released from both liposomal nanocarriers within 3 h, indicative of both a relatively rapid release mechanism and a lack of effect of surface-bound InvA497 on release kinetics. This burst release of gentamicin was not expected to have an adverse impact on testing of nanocarrier systems in efficacy studies however, as the employment of relatively shorter liposome-cell incubation periods was anticipated.
4.2. Anti-infective efficacy studies
As mentioned previously, while the strategy of using actively-targeted nanocarriers to facilitate translocation of incorporated actives across biological membranes is widely discussed in literature, such a strategy is more commonly employed in the context of tumor therapy37,38 than for intracellular delivery of anti-infectives. The use of invasin-functionalized nanocarriers for intracellular anti-infective delivery is also not a commonly employed strategy – as has been referred to earlier, the majority of literature to date reports on the potential of invasin to improve uptake of unloaded carrier systems, with efficacy assessment of invasin-functionalized carriers loaded with any form of active being seldom undertaken.18,39 The use of InvA497-functionalized liposomes for the intracellular delivery of gentamicin was therefore considered to be a strategy of great interest for exploration; it was also deemed of the utmost importance to investigate the ability of I-GL to facilitate effective intracellular delivery of gentamicin, following thorough characterization of the properties of this nanocarrier system. For this purpose, HEp-2 cells infected with the enteroinvasive bacteria S. enterica or Y. pseudotuberculosis were employed. The use of these two invasive bacteria allowed for testing of the ability of I-GL to access various intracellular compartments: once inside cells, Y. pseudotuberculosis remains within vacuoles,40 whereas S. enterica intracellular infection is characterized by both vacuolar and cytosolic replication.41An optimized bacterial invasion protocol was first established, in order to determine an MOI level and durations of both bacterial incubation and extracellular bacterial killing which would not result in an overwhelming intracellular bacterial load (Fig. S2,† and 5). Of interest and importance to note is the use of gentamicin in solution in the context of this initial bacterial invasion step, in order to exclusively kill any extracellularly located bacteria, rather than as a test formulation in the subsequent efficacy studies themselves. The employment of such a ‘gentamicin protection assay’42 has found common usage in invasion protocols,43 due to the fact that bacteria which have successfully invaded mammalian cells are ‘protected’ from the poorly permeable free drug, while extracellularly-located bacteria are not.12,44 Such a poor permeability was indeed confirmed in the subsequent efficacy testing stage of the current study: no killing of intracellular bacteria was noted to occur in the untreated control group, in which cells had been pre-treated with gentamicin solution for extracellular bacterial killing, followed by incubation with drug- and liposome-free binding buffer. A variation on this control, in which cells were pre-treated with gentamicin solution followed by incubation with gentamicin-containing binding buffer, also resulted in a negligible reduction of intracellular bacteria (data not shown). Gentamicin was therefore considered as an ideal cargo for efficacy testing of the current functionalized nanocarrier.
In the context of the efficacy testing studies themselves, HEp-2 cells containing either S. enterica or Y. pseudotuberculosis were treated with I-GL or GL, in order to investigate the impact of liposome functionalization on intracellular delivery and activity of gentamicin. In both S. enterica-infected (Fig. 6A) and Y. pseudotuberculosis-infected (Fig. 6B) HEp-2 cells, treatment with I-GL nanocarriers was seen to affect a significantly greater level of bacterial killing than treatment with GL nanocarriers. Such an observation is consistent with the previously observed greater uptake of invasin-functionalized liposomes by HEp-2 cells,20 in comparison to liposomes without surface-bound invasin. A greater effect was in fact expected as a result of treatment with GL, as gentamicin-loaded liposomes in the absence of functionalization have previously been shown to be effective in the killing of intracellular bacteria; however, this effective action has generally been seen in infected phagocytic cell models, such as macrophages.35 As demonstrated in the current work, entry of non-functionalized liposomal nanocarriers into non-phagocytic, epithelial cells appears to present a much greater challenge, and therefore requires the presence of further invasive moieties.
The noted ability of I-GL nanocarriers to reduce the intracellular bacteria load in infected cells indicates that InvA497-functionalized liposomes are not only able to invade into epithelial cells, but are also able to release their poorly-permeable payload once inside cells in order to exert a bactericidal effect. Furthermore, the effective killing of both S. enterica (cytoplasmic/vacuolar bacterium) and Y. pseudotuberculosis (vacuolar bacterium) demonstrates that gentamicin contained within the invasin-functionalized liposomal nanocarriers is able to be transported across both vacuolar membranes in order to reach bacteria enclosed within these sub-cellular compartments, and is also able to gain access to the cytosol. This is a promising observation, and could form the basis of future mechanistic studies as well as further investigations to determine the cell–liposome incubation time resulting in maximal reduction in infection load. An extension of the incubation period of Yersinia-infected cells with I-GL from 1 to 2 h has already been shown to result in a greater bacterial killing,20 an encouraging observation for conduction of further optimization studies.
5. Conclusions
In this study, liposomal nanocarriers loaded with gentamicin were successfully prepared via three different methods: the LFH method, the MCV method and the ASL method. All three methods resulted in liposome preparations exhibiting a homogenous size of approximately 200 nm and a negative surface charge. Of these three types of gentamicin-loaded liposomes, LFH liposomes showed the highest encapsulation efficiency and stability and were therefore used further to prepare InvA497-functionalized, gentamicin-containing liposomes. Surface-functionalized gentamicin liposomes were able to reduce the infection load of both S. enterica and Y. pseudotuberculosis in infected epithelial cells, meaning that such a nanocarrier was successful not only in delivering gentamicin across epithelial cell membranes, but also into vacuolar compartments. This bacteriomimetic nanocarrier system therefore represents a promising approach for intracellular delivery of poorly permeable anti-infective drugs.Acknowledgements
The authors would like to thank Dr Diana Peckys from the Leibniz-Institute for New Materials GmbH (INM), Saarbruecken, Germany and Dr Chiara De Rossi (HIPS) for help with imaging experiments (see ESI†).References
- E. M. Wellington, A. B. Boxall, P. Cross, E. J. Feil, W. H. Gaze, P. M. Hawkey, A. S. Johnson-Rollings, D. L. Jones, N. M. Lee, W. Otten, C. M. Thomas and A. P. William, Lancet Infect. Dis., 2013, 13, 155–165 CrossRef CAS PubMed.
- R. K. Ernst, T. Guina and S. I. Miller, J. Infect. Dis., 1999, 179(suppl. 2), S326–S330 CAS.
- E. Briones, C. I. Colino and J. M. Lanao, J. Controlled Release, 2008, 125, 210–227 CrossRef CAS PubMed.
- N. Abed and P. Couvreur, Int. J. Antimicrob. Agents, 2014, 43, 485–496 CrossRef CAS PubMed.
- L. Zhang, D. Pornpattananangku, C. M. Hu and C. M. Huang, Curr. Med. Chem., 2010, 17, 585–594 CrossRef CAS PubMed.
- R. Schiffelers, G. Storm and I. Bakker-Woudenberg, J. Antimicrob. Chemother., 2001, 48, 333–344 CrossRef CAS PubMed.
- Z. Drulis-Kawa and A. Dorotkiewicz-Jach, Int. J. Pharm., 2010, 387, 187–198 CrossRef CAS PubMed.
- E. Haltner, J. H. Easson and C. M. Lehr, Eur. J. Pharm. Biopharm., 1997, 44, 3–13 CrossRef CAS.
- A. Kochut and P. Dersch, Eur. J. Pharm. Biopharm., 2013, 84, 242–250 CrossRef CAS PubMed.
- R. R. Isberg and J. M. Leong, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 6682–6686 CrossRef CAS.
- P. Dersch and R. R. Isberg, Infect. Immun., 2000, 68, 2930–2938 CrossRef CAS PubMed.
- F. Uliczka, F. Pisan, J. Schaake, T. Stolz, M. Rohde, A. Fruth, E. Strauch, M. Skurnik, J. Batzilla, A. Rakin, J. Heesemann and P. Dersch, PLoS Pathog., 2011, 7, e1002117 CAS.
- P. Dersch and R. R. Isberg, EMBO J., 1999, 18, 1199–1213 CrossRef CAS PubMed.
- Z. A. Hamburger, M. S. Brown, R. R. Isberg and P. J. Bjorkman, Science, 1999, 286, 291–295 CrossRef CAS PubMed.
- S. E. Autenrieth and I. B. Autenrieth, Int. J. Med. Microbiol., 2008, 298, 69–77 CrossRef CAS PubMed.
- O. T. Bühler, C. A. Wiedig, Y. Schmid, G. A. Grassl, E. Bohn and I. B. Autenrieth, Infect. Immun., 2006, 74, 4322–4329 CrossRef PubMed.
- N. Hussain and A. T. Florence, Pharm. Res., 1998, 15, 153–156 CrossRef CAS.
- G. F. Dawson and G. W. Halbert, Pharm. Res., 2000, 17, 1420–1425 CrossRef CAS.
- A. Wiedemann, S. Linder, G. Grassl, M. Albert, I. Autenrieth and M. Aepfelbacher, Cell. Microbiol., 2001, 3, 693–702 CrossRef CAS PubMed.
- H. I. Labouta, S. Menina, A. Kochut, S. Gordon, R. Geyer, P. Dersch and C. M. Lehr, J. Controlled Release, 2015, 220, 414–424 CrossRef CAS PubMed.
- A. D. Bangham, M. M. Standish and J. C. Watkins, J. Mol. Biol., 1965, 13, 238–252 CrossRef CAS PubMed.
- T. Nii and F. Ishii, Int. J. Pharm., 2005, 298, 198–205 CrossRef CAS PubMed.
- B. Ceh and D. D. Lasic, J. Colloid Interface Sci., 1997, 185, 9–18 CrossRef CAS PubMed.
- D. Zucker, D. Marcus, Y. Barenholz and A. Goldblum, J. Controlled Release, 2009, 139, 73–80 CrossRef CAS PubMed.
- D. Sehgal and I. K. Vijay, Anal. Biochem., 1994, 218, 87–91 CrossRef CAS PubMed.
- J. Gubernator, Z. Drulis-Kawa and A. Kozubek, Int. J. Pharm., 2006, 327, 104–109 CrossRef CAS PubMed.
- Z. Zhang and S. S. Feng, Biomaterials, 2006, 27, 4025–4033 CrossRef CAS PubMed.
- S. Papadimitriou and D. Bikiaris, J. Controlled Release, 2009, 138, 177–184 CrossRef CAS PubMed.
- E. Pastor, E. Matveeva, A. Valle-Gallego, F. M. Goycoolea and M. Garcia-Fuentes, Colloids Surf., B, 2011, 88, 601–609 CrossRef CAS PubMed.
- P. Panwar, B. Pandey, P. C. Lakhera and K. P. Singh, Int. J. Nanomed., 2010, 9, 101–108 Search PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef PubMed.
- X. Xu, M. A. Khan and D. J. Burgess, Int. J. Pharm., 2012, 423, 410–418 CrossRef CAS PubMed.
- T. S. Kashi, S. Eskandarion, M. Esfandyari-Manesh, S. M. Marashi, N. Samadi and S. M. Fatemi, Int. J. Nanomed., 2012, 7, 221–234 Search PubMed.
- E. J. Ruijgroka, A. G. Vultob and E. W. van Etten, J. Liposome Res., 1999, 9, 291–300 CrossRef.
- P. Lutwyche, C. Cordeiro, D. J. Wiseman, M. St-Louis, M. Uh, M. J. Hope, M. S. Webb and B. B. Finlay, Antimicrob. Agents Chemother., 1998, 42, 2511–2520 CAS.
- C. Mugabe, A. O. Azghani and A. Omri, J. Antimicrob. Chemother., 2005, 55, 269–271 CrossRef CAS PubMed.
- A. Jhaveri and V. Torchilin, Expert Opin. Drug Delivery, 2015, 1–22 Search PubMed.
- F. Marcucci and F. Lefoulon, Drug Discovery Today, 2004, 9, 219–228 CrossRef CAS PubMed.
- E. G. Ragnarsson, I. Schoultz, E. Gullberg, A. H. Carlsson, F. Tafazoli, M. Lerm, K. E. Magnusson, J. D. Söderholm and P. Artursson, Lab. Invest., 2008, 88, 1215–1226 CrossRef CAS PubMed.
- M. N. Starnbach and M. J. Bevan, J. Immunol., 1994, 153, 1603–1612 CAS.
- P. Malik-Kale, S. Winfree and O. Steele-Mortimer, PLoS One, 2012, 7, e38732 CAS.
- J. M. Leong, R. S. Fournier and R. R. Isberg, EMBO J., 1990, 9, 1979–1989 CAS.
- E. A. Elsinghorst, Methods Enzymol., 1994, 236, 405–420 CAS.
- J. Shi and J. E. Casanova, Mol. Biol. Cell, 2006, 17, 4698–4708 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02988d |
| ‡ Current address: Department of Chemistry, Faculty of Science & Cellular and Molecular Bioengineering Research Laboratory (CMBRL), Schulich School of Engineering, University of Calgary, Calgary, Canada. |
| This journal is © The Royal Society of Chemistry 2016 |