
An ultrasensitive sandwich-type electrochemical immunosensor based on functionalized mesoporous carbon for IgG detection
Yuying
Yang
a,
Qing
Liu
*a,
Yan
Liu
a,
Zhaohui
Li
c,
Yueyun
Li
a,
Ping
Wang
a,
Daopeng
Zhang
a,
Zengdian
Zhao
b,
Yuying
Liu
a and
Yunhui
Dong
*a
aSchool of Chemical Engineering, Shandong University of Technology, Zibo, 255049, P. R. China. E-mail: qingliu0315@163.com; dyh651118@126.com; Fax: +86-533-2781664; Tel: +86-533-2781664
bAnalysis and Testing Center, Shandong University of Technology, Zibo, 255049, P. R. China
cDepartment of Energy and Engineering, Dongying Vocational College of Science & Technology, China
First published on 15th March 2016
Abstract
Immunoglobulin (IgG) plays an important role in the accurate diagnosis of some diseases. Conventional clinical detection is expensive and has low sensitivity. To overcome these disadvantages, in this study, a novel and ultrasensitive electrochemical immunosensor based on functionalized ordered mesoporous carbons (OMC) was developed. To this purpose, the mesoporous carbon was functionalized with 3-aminopropyl triethoxysilane to stabilize gold nanoparticles (Au NPs) for the efficient adsorption of chromium ions (Cr3+) and secondary antibodies (Ab2). The prepared immunoassay is better than conventional label-free electrochemical immunoassays due to the synergetic effect provided by the Cr3+@Au@OMC. The prepared nanocomposites (Cr3+@Au@OMC) functioned as a signal magnification label, as it has superior electrocatalytic activity for hydrogen peroxide reduction. Meanwhile, Au NPs were electrodeposited on the electrode surface to increase the amount of primary antibodies (Ab1) being absorbed. Under the optimal conditions, a linear range was identified from 0.01 pg mL−1to 100 ng mL−1 with a detection limit of 3.33 pg mL−1, comparing favorably with the conventional limit of 3 ng mL−1. Thus, the developed immunosensor has great potential in clinical and diagnostic applications due to its high sensitivity, good selectivity and stability for the quantitative detection of IgG.
1. Introduction
Recently, virus or disease outbreaks are occurring more frequently worldwide, in particular from communicable and infectious viruses such as the pandemics of H1N1 and measles in the regions of Eastern and Southern Africa.1 Early detection is important to prevent the wide spread of such diseases. However, on-site analytical tools are usually not sufficient to ensure early or timely detection. Thus, there is an urgent requirement for the development of analytical devices for disease, or early detection or surveillance methods for viruses. Immunoglobulin (Ig) is an important component in the immune systems of living bodies, and the level of Ig is frequently used as a detection method to recognize bacteria and viruses.2There are five types of Igs in human plasma, among which human immunoglobulin G (IgG) is the most important, accounting for about 75% of the total Igs.3,4 The most distinguishable feature of IgG is that it is able to identify and help antigens to remove toxic and pathogenic agents by the formation of a new compound, which is then engulfed by phagocytes5. Therefore, the detection of IgG in human serum is an important part of clinical chemistry.
Many electrochemical immunosensors have been developed and used for the detection of HIgG.6,7 Compared with conventional immunoassays, such as enzyme linked immunosorbent assays (ELISA),8 fluoroimmunoassays,9 and chemiluminescence immunoassays,10 electrochemical immunoassays have been received increasing attention in food safety supervision,11,12 environmental monitoring,13,14 and clinical diagnostics15,16 because of their unique advantages, such as low cost, high sensitivity, quick analysis and miniaturization.13,17–19
A variety of nanomaterials are used for the fabrication of immunosensors to achieve signal amplification, which can improve the sensitivity of electrochemical analysis.20,21 Among these efforts, ordered mesoporous carbon (OMC) materials have good potential in the fabrication of electrochemical biosensors because of their favorable properties, such as uniform and tailored pore structure, high specific surface area, good biocompatibility and superior electrical conductivity.22–24 For example, Ndamanisha and Guo proposed an electrochemical detection scheme for morphine utilizing a ordered mesoporous carbon modified glassy carbon electrode.25 Li et al. reported that ferrocenecarboxylic acid functionalized ordered mesoporous carbon can be applied to the determination of UA content in urine samples.26 The determination of catechol on ordered mesoporous carbon modified electrodes has also been reported.27 However, the poor film forming properties of OMC might influence the performance of the biosensor.
In recent years, the incorporation of heteroatoms has received increasing attention: for example, the doping strategy can be used to enhance the optical, electrical, semiconducting and surface properties of OMC. Due to the particular properties of OMC, doping OMC with gold nanoparticles (Au NPs) is a topic of vital interest. Au NPs have been widely used for the construction of electrochemical immunosensors,28 as they can promote the redox behavior of H2O2 and facilitate antibody immobilization.29 The surface functionalization of OMC with ion adsorption would provide extra capability to intensify the chemical modification.30,31 The adsorbed Cr3+ can further promote the redox behavior of H2O2, which can be used to amplify the detection signal.
In this research, a novel sandwich-type electrochemical immunosensor was designed to detect IgG. The synergetic effect presented by gold nanoparticles functionalized OMC loaded with chromium ions (Cr3+@Au@OMC) was employed to increase the sensitivity of the immunosensor towards the reduction of H2O2. Electroplated Au NPs were adsorbed on the modified electrode to immobilize primary antibodies (Ab1) which have amino groups (–NH2).32,33 The proposed novel immunosensor strategy provides a useful technology for the detection of IgG in human serum due to its high current response, relatively wide linear range and low detection limit.
2. Materials and methods
2.1. Reagents and apparatus
Human immunoglobulin G (IgG) and goat anti-human IgG were purchased from Dingguochangsheng Biotechnology LLC. (Beijing, China). HAuCl4·4H2O was purchased from Sigma-Aldrich Co., Ltd. (Beijing, China). Bovine serum albumin (BSA, 96–99%) was purchased from Sigma Reagent Co., Ltd. (St. Louis, MO, USA). 3-Aminopropyl triethoxysilane was purchased from XiBao Biological Technology Co., Ltd. (Shanghai, China). K3[Fe(CN)6] was obtained from Sinopharm Chemical Reagent Co., Ltd. (Beijing, China). A mixed Na2HPO4 and KH2PO4 stock solution was prepared to form phosphate buffered saline (PBS, pH = 7.4) that was used as the electrolyte in the process of the electrochemical measurements. All the experimental processes used ultrapure water (18.25 MΩ cm, 24 °C). All accessible chemicals and solvents were analytical grade and used without further purification. The prepared solutions were maintained at 4 °C prior to use.2.2. Preparation of the amino-functionalized OMC
OMC (0.1 g) and 3-aminopropyl triethoxysilane (0.1 mL) were mixed in anhydrous ethanol (10 mL). The mixture was heated to 70 °C and further refluxed for 1.5 h in an oil bath. Subsequently, the mixture was cooled to room temperature and separated. Finally, the prepared amino-functionalized OMC was stored at 50 °C.2.3. Preparation of Au@OMC
Au NPs were prepared according to the classical method using sodium citrate to reduce AuCl4− ions.34 Briefly, HAuCl4 (0.01 wt%, 100 mL) was refluxed and heated to boiling; then sodium citrate (1.5 mL, 10 mg mL−1) was added to a solution which had been stirred for 15 min. Finally, a wine-red solution was obtained, indicating the formation of Au NPs. The resulting solution was cooled to room temperature and stored at 4 °C for further usage.The prepared amino-functionalized OMC (10 mg) was mixed with the Au NPs solution (40 mL) and stirred for 12 h. The Au NPs were adsorbed on surface of the OMC through electrostatic interaction. The obtained material was dried and referred as Au@OMC.
2.4. Preparation of Cr3+@Au@OMC
A solution containing Au@OMC (4 mg) was mixed with chromium nitrate solution (4 mL, 1 mg mL−1) with oscillation for 24 h to ensure that Cr3+ was fully absorbed onto the Au@OMC. The Cr3+@Au@OMC sample was obtained by centrifugation.2.5. Preparation of Cr3+@Au@OMC/Ab
Cr3+@Au@OMC (2 mg mL−1) was added to an equal volume of secondary antibodies (Ab2) (10 μg mL−1). The mixture was stored in a shaking incubator at 4 °C for 12 h. Due to the interaction between Au NPs and –NH2 on the antibodies, Ab2 (protein) could be effectively conjugated onto the Cr3+@Au@OMC.32,33 The Cr3+@Au@OMC/Ab2 was collected after centrifugation and configured into different concentrations, then stored at 4 °C for future use. The procedure of Cr3+@Au@OMC/Ab2 preparation is outlined in Fig. 1A.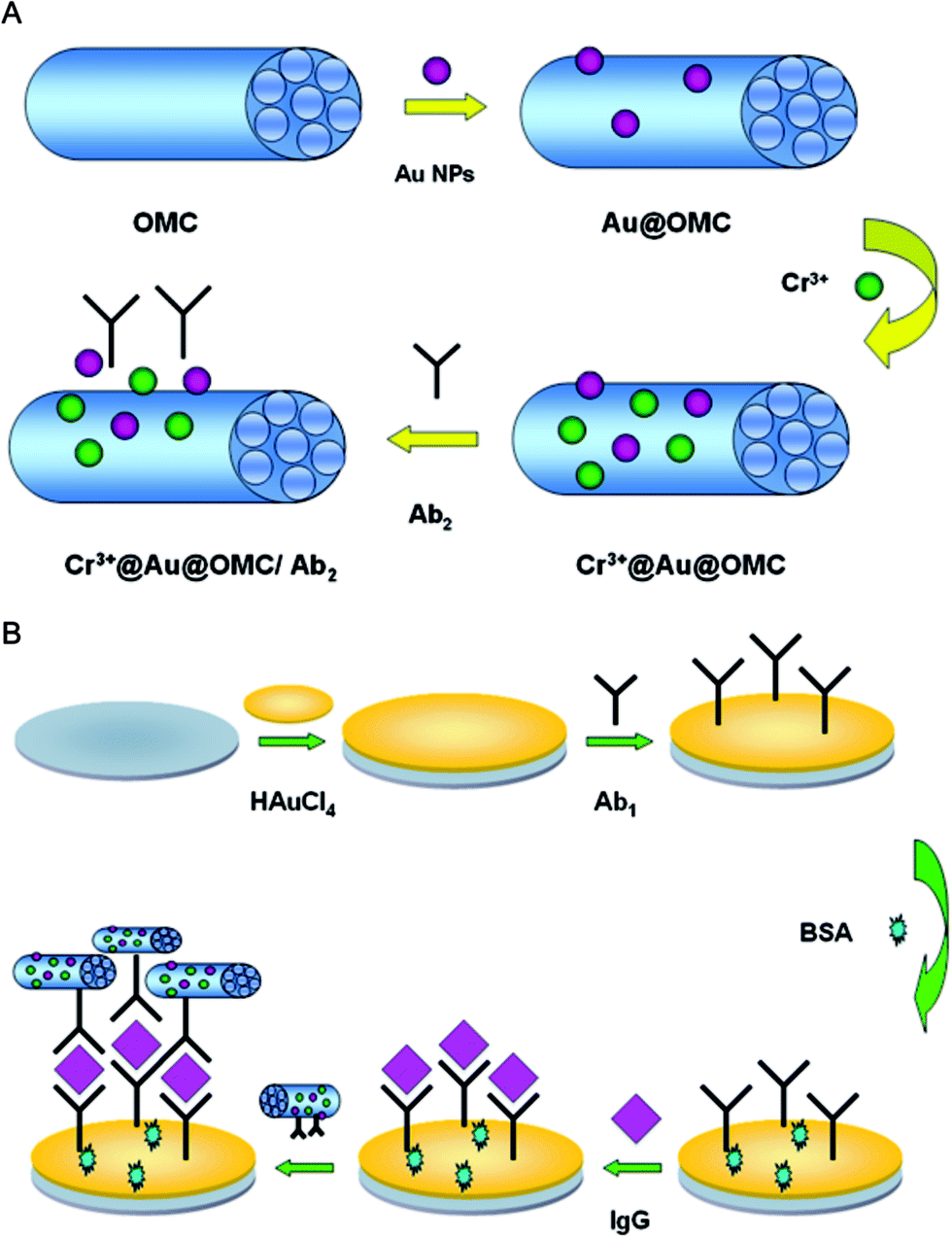 | ||
| Fig. 1 (A) The preparation procedure of Cr3+@Au@OMC/Ab2. (B) Schematic of the sandwich-type electrochemical immunosensor. | ||
2.6. Fabrication of the sandwich-type immunosensor
The process for the construction of the sandwich type electrochemical immunosensor is depicted in Fig. 1B. Generally, prior to modification, bare glassy carbon electrode (GCE) was polished using alumina polishing powders with particle sizes of 1.0, 0.3, and 0.05 μm, respectively, followed by sonication in ultrapure water and desiccation under a high-purity N2 stream to obtain a mirror surface. The electrodeposition of Au NPs on GCE was performed in HAuCl4 (1 wt%, 2 mL) aqueous solution at a constant potential of −0.2 V for 30 s.35 Ab1 (10 μg mL−1, 6 μL) was dropped onto the resulting Au NPs/GCE and incubated at 4 °C until dried. After washing, in order to eliminate nonspecific binding sites, BSA (1 wt%, 3 μL) solution was dropped onto the electrode and incubated for 0.5 h. Subsequently, the modified electrode was thoroughly cleaned with ultrapure water to remove the non-chem-adsorbed species. The resulting electrode was incubated with varying concentrations of IgG (1 × 10−6 to 1 × 102 ng mL−1, 6 μL) for 1 h at room temperature, after which the electrode was thoroughly washed with ultrapure water to remove unbound IgG molecules. Then the prepared Cr3+@Au@OMC/Ab2 solution (2 mg mL−1, 6 μL) was dropped onto the surface of the modified electrode for the immune reaction. Finally, the prepared electrode was thoroughly rinsed again with ultrapure water to remove all the physically absorbed species.2.7. Experimental measurements
All electrochemical experiments were performed using a conventional three-electrode system: a GCE 4 mm in diameter as the working electrode, a saturated calomel electrode (SCE) as the reference electrode, and a platinum wire electrode as the counter electrode. All cyclic voltammetry experiments (CVs) were performed in the conventional electrochemical cell by scanning the potential from −1.0 V to 1.0 V. An amperometric i–t curve was used to record the electrochemical signal by scanning the potential at −0.4 V. 5.0 M (10 μL) H2O2 was added to the PBS (10 mL, pH = 7.4) after the current stabilized under stirring. In order to characterize the assembly process of the modified electrode, the Nyquist plots of AC impedance spectroscopy were recorded in a solution containing 0.1 M KCl and 2.5 mM Fe(CN)63−/Fe(CN)64−.3. Results and discussion
3.1. Characterization of OMC and Au@OMC composites
Scanning electron microscopy (SEM) was used to characterize the morphologies of the OMC and Au@OMC composites. As can be seen from the SEM image (Fig. 2A), the external structure of untreated OMC is a short column-like morphology with uniform pore size distribution. Comparatively, many small particles were attached on the OMC after Au NPs adsorption, which clearly proves the presence of Au NPs attached on the surface of the OMC by physical adsorption (Fig. 2B). The Energy Dispersive X-ray spectroscopy (EDX) spectrum further confirms the presence of Au in the composition. | ||
| Fig. 2 (A) SEM image of OMC; (B) Au@OMC; (C) EDX spectrum of Au@OMC. | ||
3.2. Characterization of Cr3+@Au@OMC
In order to characterize the electrocatalytic properties of the nanomaterials, electroplated Au NPs, Au NPs, OMC, Au@OMC and Cr3+@Au@OMC were modified onto bare GC electrodes, respectively, for electrocatalytic H2O2 reduction. As shown in Fig. 3A, in the amperometric i–t curve, a weak signal was detected when electroplated Au NPs (curve a), Au NPs (curve b) and OMC (curve c) were modified onto the electrodes. When Au@OMC (curve d) was applied as the label, the electrochemical signal was magnified, which is attributed to the synergistic effects of OMC and Au NPs. As expected, the immunosensor using Cr3+@Au@OMC as a signal enhancer displayed the highest current change (curve e). Thus, it could be deduced that the redox procedure of H2O2 can be promoted by the redox procedure of Cr3+. These results clearly show that Cr3+@Au@OMC exhibited superior potential for immunosensor applications compared to the other nanomaterials being tested.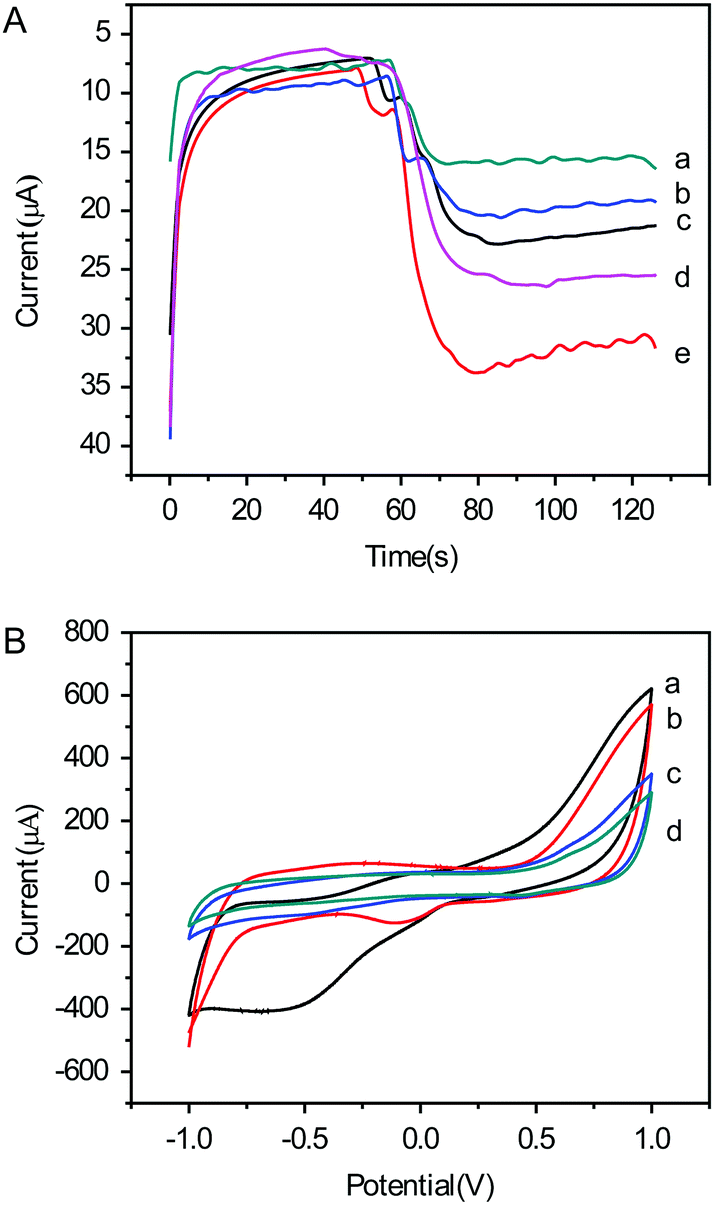 | ||
| Fig. 3 (A) Amperometric response of the immunosensors with different nanomaterials: (a) electroplating Au NPs, (b) Au NPs, (c) OMC, (d) Au@OMC, (e) Cr3+@Au@OMC. (B) CVs of the immunosensor: using Cr3+@Au@OMC as transducing material in PBS at pH = 7.4 after (curve a) and before (curve b) the addition of 5.0 mM H2O2; using Au@OMC as transducing material after (curve c) and before (curve d) the addition of 5.0 mM H2O2. | ||
CV results further confirmed the excellent catalytic activity of Cr3+@Au@OMC. As shown in Fig. 3B, there was no electrochemical signal at the Au@OMC modified GC electrode (curve d) in PBS at pH = 7.4. After the addition of 5.0 mM H2O2, a slight cathodic current was observed, attributed to H2O2 reduction (curve c). Comparatively, a reduced peak was observed around −0.2 V in PBS using Cr3+@Au@OMC as the transducing material (curve b), which could be ascribed to the reduction of Cr3+. These observations obviously show that Cr3+ was adsorbed successfully on Au@OMC. A significant increase of the cathodic current and a decrease of the anodic current around the potential of 0.4 V were detected upon the addition of 5.0 mM H2O2 (curve a), indicating that Cr3+@Au@OMC has good electrocatalytic performance in H2O2 reduction.
3.3. Optimization of synthesis conditions
The catalytic activity of Cr3+@Au@OMC was further investigated at different pH values of the PBS and under different concentrations of Cr3+@Au@OMC to optimize the experimental conditions. The effects of different pH values of PBS were investigated under the same concentration of Cr3+@Au@OMC (2.5 mg mL−1) modified on the surface of GCE. As shown in Fig. 4A, the current signal increased from pH 5.8 to 8.4, which may be because the change in pH value could influence the electrocatalytic process towards the reduction of H2O2 and the activity of biological proteins.36 These results indicate that the optimal electrocatalytic current response was achieved at pH = 7.4. Therefore, PBS at pH = 7.4 was chosen as the electrolyte for further electrochemical performance tests. | ||
| Fig. 4 (A) The optimal experimental conditions of pH. (B) The optimal experimental conditions of Cr3+@Au@OMC concentration. Error bars show RSD (n = 5). | ||
The concentration of the Cr3+@Au@OMC solution used to coat the surface of GC electrode was further investigated in PBS at pH = 7.4. As shown in Fig. 4B, with the increment of the concentration from 0.5 to 4.0 mg mL−1, the current signal initially improved to a maximum value at a concentration of 2.5 mg mL−1 and then decreased. It can be inferred that the interface electron transfer resistance caused by the increase of Cr3+@Au@OMC film thickness makes the electron transfer more difficult.37 Therefore, a Cr3+@Au@OMC concentration of 2.5 mg mL−1 was used as the optimal concentration in this study.
3.4. Characterization of the immunosensor
Electrochemical impedance spectroscopy (EIS) is regarded as a useful method to monitor the changes in an assembly process on the surface of an electrode.38 Impedance spectroscopy comprises two parts: a linear part and a semicircle part. The linear part at low frequencies is associated with electrochemical behavior limited by diffusion. The semicircle part at high frequencies is associated with the electrochemical process subject to electron transfer, where the diameter corresponds to the resistance.39 The Nyquist plots of EIS were recorded from 0.1 to 105 Hz at 0.18 V in a solution containing 0.1 M KCl and 2.5 mM Fe(CN)63−/Fe(CN)64− (Fig. 5A). The bare GCE exhibited a smaller diameter with a resistance value of 143.8 Ω (curve a). When Au NPs were electrodeposited on the surface of bare GCE (curve b), the resistance value (106.8 Ω) was much smaller because the electrodeposited Au NPs promoted electron transfer. After incubation with Ab1, the resistance value was further increased to 257.6 Ω, which indicates that Ab1 was immobilized on the electrode successfully and blocked electron transfer (curve c). Additionally, when the BSA was modified onto the electrode surface, the resistance was further increased to 318.2 Ω (curve d). This high resistance is due to the blocking effect of electron transfer by modified protein molecules on the surface of the electrode. Similarly, the resistance was significantly enlarged to 519.3 Ω (curve e) when IgG was applied onto the electrode. In contrast, when Cr3+@Au@OMC/Ab2 was modified on the electrode, an obvious decrease in the resistance value (372.7 Ω) could be observed (curve f), suggesting that Cr3+@Au@OMC/Ab2 would promote the electron transfer. | ||
| Fig. 5 (A) Nyquist plots recorded from 1 to 105 Hz in the presence of 2.5 mM Fe(CN)63−/Fe(CN)64− containing 0.1 M KCl and at bare GCE (a), Au NPs/GCE (b), Ab1/Au NPs/GCE (c), BSA/Ab1/Au NPs/GCE (d), IgG/BSA/Ab1/Au NPs/GCE (e) and Cr3+@Au@OMC-Ab2/IgG/BSA/Ab1/Au NPs/GCE (f). (B) Electrochemical impedance plots of the immunosensor for the determination of different concentrations of IgG at −0.4 V in PBS containing 5.0 mM H2O2: (a) 3.3 × 106 ng mL−1, (b) 10−5 ng mL−1, (c) 10−4 ng mL−1, (d) 10−3 ng mL−1, (e) 10−2 ng mL−1, (f) 10−1 ng mL−1, (g) 1 ng mL−1, (h) 10 ng mL−1 and (i) 102 ng mL−1. (C) Calibration curve of the immunosensor toward different concentrations of IgG. Error bars show RSD (n = 5). | ||
Under the optimal conditions, a sandwich-type electrochemical immunosensor using gold electroplating to amplify the signal and Cr3+@Au@OMC/Ab2 as the label was used to determine different concentrations of IgG by amperometric i–t curve in PBS at −0.4 V. The current–time response towards the reduction of 5.0 mM H2O2 under different IgG concentrations is shown in Fig. 5B, and the relationship between the amperometric response and the logarithmic value of the IgG concentration is displayed in Fig. 5C. A linear calibration curve was obtained with IgG concentrations ranging from 0.01 pg mL−1 to 100 ng mL−1, and the limit of detection (LOD) calculated was 3.3 fg mL−1 at a signal to noise ratio (S/N) of 3. The regression equation of the calibration curve is I = 2.32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logC + 18.82, with a correlation coefficient of 0.9976. The low detection limit is attributed to the following factors: good biocompatibility of electroplating Au NPs used to capture more Ab1, the large surface area of Cr3+@Au@OMC conjugated to large amounts of Ab2, and the superior electrocatalytic activity and fast electron transfer of Cr3+@Au@OMC.
logC + 18.82, with a correlation coefficient of 0.9976. The low detection limit is attributed to the following factors: good biocompatibility of electroplating Au NPs used to capture more Ab1, the large surface area of Cr3+@Au@OMC conjugated to large amounts of Ab2, and the superior electrocatalytic activity and fast electron transfer of Cr3+@Au@OMC.
3.5. Reproducibility, selectivity and stability of the immunosensor
The selectivity, reproducibility and stability of a fabricated immunosensor are three prerequisite conditions for practical application considerations;40 thus, they were investigated in this work.Four different electrodes were prepared for parallel detection of 10 ng mL−1 IgG to evaluate the reproducibility of the immunosensor. As shown in Fig. 6A, the relative standard deviation (RSD) was only 1.8%, indicating that the proposed immunoassay displays good precision and reproducibility.
 | ||
| Fig. 6 (A) Amperometric change response of the biosensor to four different electrodes treated in the same way. (B) Current responses of the immunosensor to 10 ng mL−1 IgG (1), 10 ng mL−1 IgG + 100 ng mL−1 vitamin C (2), 10 ng mL−1 IgG + 100 ng mL−1 glucose (3), 10 ng mL−1 IgG + 100 ng mL−1 BSA (4). Error bars show RSD (n = 5). (C) The stability study of the IgG immunosensor. | ||
Vitamin C, glucose and BSA were used as interfering substances to evaluate the selectivity of the immunosensor. IgG solution (1 ng mL−1) containing 100 ng mL−1 of interfering substances was measured by the designed immunosensor (Fig. 6B). The current signal variation was 2.1%, less than 5% of that without interference, indicating that the selectivity of the immunosensor is acceptable.
The stability of the prepared immunosensor was inspected by maintaining the electrodes at 4 °C, with the current response being tested each week. As shown in Fig. 6C, after the first week, the current response of the immunosensor only had a minor change of 1.3%. After three weeks, the current response retained 78.5% of its initial current, reflecting the good stability of the immunosensor.
3.6. Real sample analysis
In order to the evaluate the accuracy and feasibility of the proposed immunoassay for real sample analysis, the standard addition method was used to detect the recoveries of different concentrations of IgG in serum samples (Table 1). IgG solutions with concentrations of 1, 5, and 10 ng mL−1 were added to serum samples. The RSD ranged from 3.661 to 6.228% and the recovery rate was 100.0 to 101.4%. The results imply that the proposed immunosensor could be effectively applied in the clinical determination of IgG levels in serum samples.| Initial IgG concentration in sample (ng mL−1) | Added IgG concentration (ng mL−1) | Measured concentration after addition (ng mL−1) | Average value (ng mL−1) | RSD (%, n = 5) | Recovery (%, n = 5) |
|---|---|---|---|---|---|
| 0.97 | 1.00 | 2.02, 1.97, 2.04, 1.95, 2.01 | 1.998 | 3.661 | 101.4 |
| 0.97 | 5.00 | 5.99, 5.98, 6.01, 5.87, 6.03 | 5.976 | 6.228 | 100.1 |
| 0.97 | 10.00 | 10.95, 11.01, 10.99, 10.89, 11.02 | 10.972 | 5.310 | 100.0 |
4. Conclusions
An ultrasensitive sandwich-type electrochemical immunosensor based on Au NPs as the platform and Cr3+@Au@OMC as the signal label for the quantitative determination of IgG was developed. To design the high performance electrochemical immunosensor, Au NPs was electroplated on an electrode, which could effectively capture Ab1 and amplify the detection signal. The prepared Cr3+@Au@OMC nanocomposites were used to capture Ab2 and functioned as the signal magnification label for H2O2 reduction. The developed immunosensor displayed a linear response with a wide range from 0.01 pg mL−1 to 100 ng mL−1 and a low detection limit (0.33 pg mL−1), and it possessed high sensitivity, good reproducibility, stability and excellent selectivity. The designed immunosensor may provide a new and promising alternative for the accurate determination of other biomolecules in clinical diagnosis.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (21302057; 21405095; 21575079) and the Young Talents Joint Fund of Shandong Province (ZR2015JL005).References
- W. H. Organization, Global tuberculosis control: WHO report 2010, World Health Organization, 2010 Search PubMed.
- L. Liu, Y. Li, L. Tian, T. Guo, W. Cao and Q. Wei, Sens. Actuators, B, 2015, 211, 170–176 CrossRef CAS.
- J. N. Arnold, M. R. Wormald, R. B. Sim, P. M. Rudd and R. A. Dwek, Annu. Rev. Immunol., 2007, 25, 21–50 CrossRef CAS PubMed.
- L. Campanella, E. Martini and M. Tomassetti, Sens. Actuators, B, 2008, 130, 520–530 CrossRef CAS.
- S. Bozzaro, C. Bucci and M. Steinert, Int. Rev. Cell Mol. Biol., 2008, 271, 253–300 CAS.
- L.-P. Qiu, C.-C. Wang, P. Hu, Z.-S. Wu, G.-L. Shen and R.-Q. Yu, Talanta, 2010, 83, 42–47 CrossRef CAS PubMed.
- H. Qi, C. Wang and N. Cheng, Microchim. Acta, 2010, 170, 33–38 CrossRef CAS.
- E. Engvall and P. Perlmann, Immunochemistry, 1971, 8, 871–874 CrossRef CAS PubMed.
- E. Soini and I. Hemmilä, Clin. Chem., 1979, 25, 353–361 CAS.
- T. Tanaka and T. Matsunaga, Anal. Chem., 2000, 72, 3518–3522 CrossRef CAS PubMed.
- W. Jin, G. Yang, L. Wu, Q. Wang, H. Shao, A. Qin, B. Yu, D. Li and B. Cai, Food Control, 2011, 22, 1609–1616 CrossRef CAS.
- G. Zhao, F. Xing and S. Deng, Electrochem. Commun., 2007, 9, 1263–1268 CrossRef CAS.
- N. Li, Y. Wang, Y. Li, W. Cao, H. Ma, D. Wu, B. Du and Q. Wei, Sens. Actuators, B, 2014, 202, 67–73 CrossRef CAS.
- F. Zhang, S. H. Yang, T. Y. Kang, G. S. Cha, H. Nam and M. E. Meyerhoff, Biosens. Bioelectron., 2007, 22, 1419–1425 CrossRef CAS PubMed.
- Y. Cai, H. Li, B. Du, M. Yang, Y. Li, D. Wu, Y. Zhao, Y. Dai and Q. Wei, Biomaterials, 2011, 32, 2117–2123 CrossRef CAS PubMed.
- Y. Zhuo, Y.-Q. Chai, R. Yuan, L. Mao, Y.-L. Yuan and J. Han, Biosens. Bioelectron., 2011, 26, 3838–3844 CrossRef CAS PubMed.
- K. M. Mayer, S. Lee, H. Liao, B. C. Rostro, A. Fuentes, P. T. Scully, C. L. Nehl and J. H. Hafner, ACS Nano, 2008, 2, 687–692 CrossRef CAS PubMed.
- H. Tang, J. Chen, L. Nie, Y. Kuang and S. Yao, Biosens. Bioelectron., 2007, 22, 1061–1067 CrossRef CAS PubMed.
- S. Weng, M. Chen, C. Zhao, A. Liu, L. Lin, Q. Liu, J. Lin and X. Lin, Sens. Actuators, B, 2013, 184, 1–7 CrossRef CAS.
- J. Dong, H. Zhao, M. Xu, Q. Ma and S. Ai, Food Chem., 2013, 141, 1980–1986 CrossRef CAS PubMed.
- L. Zhao, S. Li, J. He, G. Tian, Q. Wei and H. Li, Biosens. Bioelectron., 2013, 49, 222–225 CrossRef CAS PubMed.
- M. Hartmann, A. Vinu and G. Chandrasekar, Chem. Mater., 2005, 17, 829–833 CrossRef CAS.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS PubMed.
- J. C. Ndamanisha and L. Guo, Biosens. Bioelectron., 2008, 23, 1680–1685 CrossRef CAS PubMed.
- F. Li, J. Song, C. Shan, D. Gao, X. Xu and L. Niu, Biosens. Bioelectron., 2010, 25, 1408–1413 CrossRef CAS PubMed.
- X. Xu, M. Guo, P. Lu and R. Wang, Mater. Sci. Eng., C, 2010, 30, 722–729 CrossRef CAS.
- X. Li, R. Yuan, Y. Chai, L. Zhang, Y. Zhuo and Y. Zhang, J. Biotechnol., 2006, 123, 356–366 CrossRef CAS PubMed.
- Q. Cao, H. Zhao, Y. Yang, Y. He, N. Ding, J. Wang, Z. Wu, K. Xiang and G. Wang, Biosens. Bioelectron., 2011, 26, 3469–3474 CrossRef CAS PubMed.
- H. Niu, R. Yuan, Y. Chai, L. Mao, H. Liu and Y. Cao, Biosens. Bioelectron., 2013, 39, 296–299 CrossRef CAS PubMed.
- S. S. Wong, A. T. Woolley, E. Joselevich, C. L. Cheung and C. M. Lieber, J. Am. Chem. Soc., 1998, 120, 8557–8558 CrossRef CAS.
- Z.-D. Gao, F.-F. Guan, C.-Y. Li, H.-F. Liu and Y.-Y. Song, Biosens. Bioelectron., 2013, 41, 771–775 CrossRef CAS PubMed.
- J. Han, Y. Zhuo, Y. Chai, Y. Yu, N. Liao and R. Yuan, Anal. Chim. Acta, 2013, 790, 24–30 CrossRef CAS PubMed.
- G. Frens, Nature, 1973, 241, 20–22 CAS.
- Y. Wu, W. Xu, L. Bai, Y. Yuan, H. Yi, Y. Chai and R. Yuan, Biosens. Bioelectron., 2013, 50, 50–56 CrossRef CAS PubMed.
- R. Yuan, D. Tang, Y. Chai, X. Zhong, Y. Liu and J. Dai, Langmuir, 2004, 20, 7240–7245 CrossRef CAS PubMed.
- S. Zhang, H. Ma, L. Yan, W. Cao, T. Yan, Q. Wei and B. Du, Biosens. Bioelectron., 2014, 59, 335–341 CrossRef CAS PubMed.
- R. Pei, Z. Cheng, E. Wang and X. Yang, Biosens. Bioelectron., 2001, 16, 355–361 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, Y. Su, F. Li, H. Ma, H. Li, B. Du and Q. Wei, Talanta, 2014, 124, 60–66 CrossRef CAS PubMed.
- A. Guo, Y. Li, W. Cao, X. Meng, D. Wu, Q. Wei and B. Du, Biosens. Bioelectron., 2015, 63, 39–46 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |