
Synthesis of curly graphene nanoribbon/polyaniline/MnO2 composite and its application in supercapacitor†
Zhixiong Yinab,
Haihui Zhou*ab,
Chaopeng Fuab,
Ningshuang Zhangab,
Dan Liuab and
Yafei Kuang*ab
aState Key Laboratory for Chemo/Biosensing and Chemometrics, Hunan University, Changsha, China. E-mail: haihuizh@163.com; Fax: +86 731 88713642; Tel: +86 731 88821603
bCollege of Chemistry and Chemical Engineering, Hunan University, Changsha, China. E-mail: yafeik@163.com; Tel: +86 731 88821863
First published on 13th April 2016
Abstract
Curly graphene nanoribbon/polyaniline/MnO2 (CGNR/PANI/MnO2) nanocomposites with a unique structure is prepared. The formation mechanism of the CGNR/PANI/MnO2 nanocomposite was proposed, and the morphology and structure were characterized by electron microscopy, X-ray diffraction, and Raman spectroscopy. The CGNR/PANI/MnO2 nanocomposite was investigated for supercapacitor applications. The CGNR/PANI/MnO2 electrode delivered a very high specific capacitance of 496 F g−1, which was much higher than that of CGNR (131 F g−1), PANI (301 F g−1) and MnO2 (33 F g−1), whereas 81.1% of its initial capacitance was retained after 5000 cycles at a scan rate of 50 mV s−1. The CGNR/PANI/MnO2 electrode was also evaluated via a two-electrode configuration, and the supercapacitor delivered a specific capacitance of 103 F g−1. The enhanced electrochemical performance of the CGNR/PANI/MnO2 electrode was ascribed to the unique structure and the synergetic effect of the three components in the composite.
Introduction
With the overconsumption of fossil fuels and the rapid deterioration of the environment, renewable and sustainable energy conversion technologies are urgently needed to address these problems.1–3 Among the various energy storage and conversion devices, the supercapacitor (also termed as electrochemical capacitor) is one of the most prominent energy storage systems due to its high power density, high energy efficiency, and long cycle life.4–7 As a result, supercapacitors have been studied extensively in hybrid electric vehicles, portable electronics, micro-autonomous robots and backup power systems.8,9Supercapacitors can be classified into two types on the basis of different charge storage mechanisms: electric double layer capacitors (EDLCs)10 and pseudocapacitors.11 EDLCs store energy at the interface between the electrodes and electrolytes;12 therefore, their capacitive performance relies strongly on the specific surface area and porosity of the electrode materials such as activated carbon, carbon nanotubes, and graphene. However, EDLCs normally display a relatively low capacitance, lying in the range 50–200 F g−1, due to the limited accessible surface area and/or the stacking or aggregation of the carbon nanomaterials.13 For example, graphene, the hottest 2D carbon material, has a theoretical specific surface area of 2675 m2 g−1, which leads to a theoretical specific capacitance of 550 F g−1.14 However, the realized surface area and capacitance of graphene are far below their theoretical values.15–17 On the other hand, carbon nanotubes (CNTs), which are particularly interesting for supercapacitor electrodes because of their unique tubular porous structures and superior electrical conductivity, favor rapid ion and electron transfer.18 However, pristine CNTs are easily intertwined, causing a large decrease in surface area. Therefore, some creative approaches have been taken to address the problem. Sharma's group synthesized curly graphene nanoribbons (CGNRs) that were composed of a partial nanotube structure and partial graphene layered morphology by cutting the CNTs transversely and longitudinally, and the resulting CGNR showed a much higher specific surface area than the pristine CNTs. More importantly, these unoriented CGNRs could readily establish a 3D conductive framework, providing sufficient space to assemble other active materials.19
Pseudocapacitor electrode materials are mostly transition metal oxides and conducting polymers, storing energy through reversible redox reactions, which occur not only at the electrode surface, but also in the bulk close to the surface of the solid electrode.20–22 Therefore, pseudocapacitor electrode materials normally show much larger theoretical capacitance.23,24 However, their rate and cycling performance are moderate due to the relatively low electrical conductivity and structure destruction during charging and discharging. Among the various pseudocapacitive materials, MnO2 stands out because of its low cost, environmentally friendliness and high theoretical specific capacitance (∼1300 F g−1).25,26 Nevertheless, the capacitance of MnO2 is difficult to fully obtain due to its low intrinsic conductivity from 10−5 to 10−6 s cm−1.27 Therefore, MnO2 with various structures and morphologies were synthesized to promote its electrochemical performance.28–30 Polyaniline (PANI), as a typical conducting polymer, has attracted increasing interests owing to its high specific capacitance, good electrical conductivity, and readily synthetic method.31–33 However, swelling and shrinkage during charging and discharging resulted in the poor cycling stability of PANI.34,35
An effective way to solve the intrinsic problems of these unitary electrode materials is to fabricate new nanocomposites that combine different types of supercapacitor electrode materials together.36–38 A variety of nanocomposite electrode materials have been prepared to improve the supercapacitor performance by combining EDLCs electrode materials with pseudocapacitive materials. Yuan et al. reported that MWCNTs coated PANI displayed a high specific capacitance of up to 560 F g−1 at 1 mV s−1 and good cycling stability.39 Xu et al. reported novel and free-standing hierarchical carbon nanofiber/graphene oxide/PANI films showing a specific capacitance of up to 450.2 F g−1 at a scan rate of 10 mV s−1.40 Shen et al. showed that the PANI/graphene/CNTs had good cycling performance of more than 91% capacitance retained after 5000 cycles.41 These studies indicated that the electrochemical performance was enhanced when the composite materials were used as electrodes.
In this study, we prepared a curly graphene nanoribbon/polyaniline/MnO2 ternary nanocomposite, in which the curly graphene nanoribbon (CGNR) was used as a 3D frame to enable rapid electron transfer, and aniline monomers were then polymerized in situ in the CGNR frame to fill the space among CGNR, and finally MnO2 nanoneedles were interspersed on the CGNR/PANI composite to pack the gap between CGNR and PANI. The results show that the CGNR/PANI/MnO2 nanocomposite displays outstanding electrochemical performance due to the remarkable synergic effects of CGNR, PANI and MnO2.
Experimental section
Synthesis of OCGNR/PANI binary composite
Oxidized curly graphene nanoribbon (OCGNR) was prepared using a modified Hummers' method.42 Briefly, MWCNTs (1 g) and sodium nitrate (0.5 g) were added in 46 mL 98% H2SO4 and the suspension was then stirred for 1 h in an ice bath. Under vigorous stirring, 5 g of KMnO4 was added to the suspension at a temperature below 10 °C. Furthermore, the temperature was increased to 35 °C and the mixture was stirred for 4 h. Subsequently, 100 mL of ultrapure water was added slowly with continual agitation, and the mixture was kept stirring for another 0.5 h. After cooling to room temperature, H2O2 (5 mL) and ultrapure water (180 mL) were added to the suspension. The suspension was washed with HCl (10 vol%), ethanol and ultrapure water until the pH of the solution was neutral and dried at about 60 °C. For the preparation of OCGNR/PANI, 50 mg of OCGNR was added to 25 mL of 1 M HCl + 2.15 mM aniline solution (mass ratio of aniline to OCGNR was 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). The mixture was sonicated for 1 h to obtain a uniform suspension. Thereafter, a designed amount of ammonium peroxydisulfate (APS) was added rapidly to the abovementioned mixture under vigorous agitation, and the solution was kept stirring overnight. The final product OCGNR/PANI was obtained by centrifugation, and then washed with ultrapure water, absolute ethanol, and hexanes. The product was dried at 60 °C under vacuum for further use.
:2). The mixture was sonicated for 1 h to obtain a uniform suspension. Thereafter, a designed amount of ammonium peroxydisulfate (APS) was added rapidly to the abovementioned mixture under vigorous agitation, and the solution was kept stirring overnight. The final product OCGNR/PANI was obtained by centrifugation, and then washed with ultrapure water, absolute ethanol, and hexanes. The product was dried at 60 °C under vacuum for further use.
Synthesis of CGNR/PANI/MnO2 ternary composite
The CGNR/PANI/MnO2 nanocomposite was prepared hydrothermally. Briefly, 80 mg of the previously prepared OCGNR/PANI and 30 mg of MnSO4·H2O were added to 40 mL H2O. After ultrasonic treatment, concentrated sulfuric acid was added dropwise under constant stirring to adjust the pH. Subsequently, 15 mg of KMnO4 dissolved in 20 mL H2O was added to the previous solution dropwise with continuous stirring. The mixture was transferred immediately to a 100 mL Teflon-lined autoclave and heated to 120 °C for 8 h. After the reaction, the autoclave was allowed to cool to room temperature. The solid was separated and washed several times with ultrapure water and dried at 60 °C in a vacuum oven. The obtained composite was labeled as OCGNR/PANI/MnO2. The as-prepared product was then reduced by hydrazine monohydrate to achieve the final product CGNR/PANI/MnO2.For comparison, CGNR/PANI was also prepared. 100 mg of the previously prepared OCGNR/PANI was dispersed and sonicated in 50 mL H2O and 0.1 mL hydrazine monohydrate was added. Afterwards, the temperature was gradually increased to 95 °C and the mixture was refluxed for 2 h. The reduced materials were filtered and washed several times with ultrapure water. The obtained product was named as CGNR/PANI.
Characterization
X-ray powder diffraction (XRD) of the samples was conducted using a Rigaku D/Max Ultima II diffractometer with Cu-Kα radiation (λ = 0.15418 nm). The morphology of the samples was characterized by scanning electron microscopy (SEM, Hitachi S-4800 Japan) and transmission electron microscopy (TEM, JEM-2100F, Japan). Raman spectroscopy was carried out using a Labram-010 (France) with 632 nm laser excitation. X-ray photoelectron spectroscopy (XPS, K-Alpha 1063) was carried out using focused monochromatized Al Kα radiation.Electrochemical measurements
Electrochemical testing of the materials was measured in a three-electrode system using a Pt sheet as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode with a CHI760C potentiostat system. The working electrodes were prepared as follows: 3 mg of the sample was dispersed in H2O to make a suspension with a concentration of 1 mg mL−1, and 20 μL of the suspension was dropped onto a glassy carbon electrode with a diameter of 5 mm. After several hours of drying at room temperature, the electrode was covered with a Nafion solution (0.5 wt%, 5 μL) and dried in air again. The electrochemical properties of the CGNR/PANI/MnO2, MnO2, PANI and CGNR were tested in 1 M H2SO4 by cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and galvanostatic charge/discharge. In a two-electrode system, the symmetric CGNR/PANI/MnO2 supercapacitor was assembled using modified glassy carbon electrodes as the positive and negative electrodes.Results and discussion
Fig. 1 presents a schematic of the formation of the CGNR/PANI/MnO2 ternary nanocomposite. First, the raw MWCNTs were cut longitudinally and unzipped transversely into oxidized curly graphene nanoribbon (OCGNR) using the modified Hummers' method. The hydrophilic OCGNR was dispersed uniformly in water, which facilitated further processing. Aniline was added to the OCGNR suspension to be adsorbed on the surface of the OCGNR. Subsequently, the adsorbed aniline monomers were oxidized in situ and polymerized with the aid of APS, resulting in the formation of the OCGNR/PANI nanocomposite. During the hydrothermal reaction, the positively charged Mn2+ ions interacted strongly with the negatively charged carboxyl and hydroxyl groups on the OCGNR surface due to electrostatic interactions or covalent chemical bonding. Second, the added KMnO4 reacted with the bonded Mn2+, leading to the formation of MnO2. At the initial stage, the nucleation occurred accompanying the redox reaction on the OCGNR surface, and the oxygen-containing functional groups acted as anchor sites for the growth of MnO2 crystal. The OCGNR/PANI could control the growth of MnO2, limiting the expansion of MnO2 crystal. Finally, the OCGNR/PANI/MnO2 was reduced by hydrazine to produce the final product CGNR/PANI/MnO2.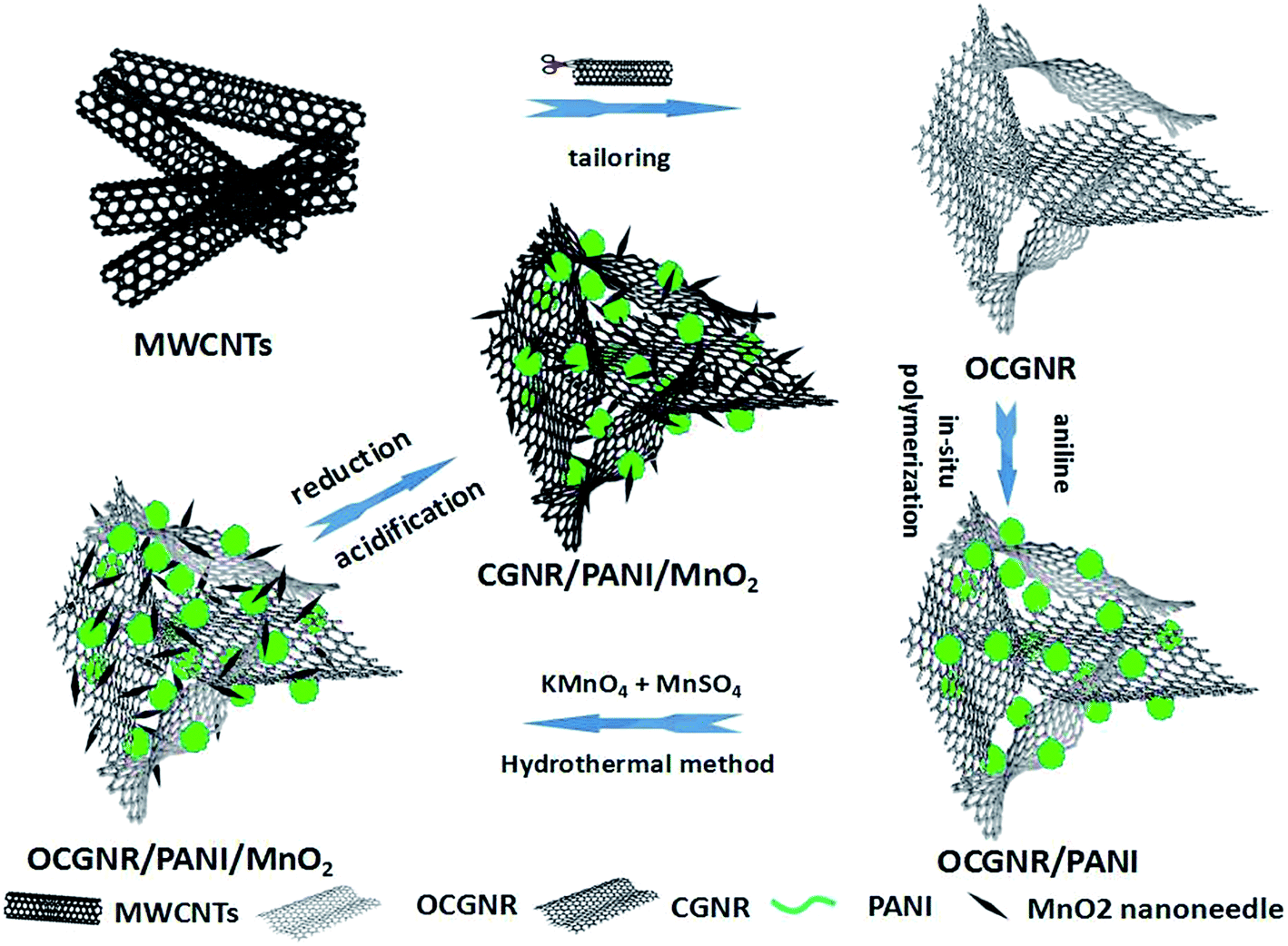 | ||
| Fig. 1 Schematic of the preparation of the CGNR/PANI/MnO2 composite. | ||
Morphology and structure
Fig. 2 shows SEM images of the MWCNTs, CGNR, CGNR/PANI and CGNR/PANI/MnO2. Fig. 2a shows the pristine MWCNTs with a diameter of 60–100 nm and a length of 5–15 μm. Fig. 2b shows the SEM image of the CGNR, and it clearly displayed that the MWCNTs were opened and unzipped into curly graphene structure. This transformation caused an increase in diameter from 100 nm to 200 nm, and this opened and wider morphology was beneficial for ions to access. Fig. 2c shows an SEM image of the CGNR/PANI. The in situ polymerized PANI with a nanoflake structure was grown on the interweaved CGNR, and the nano-PANI flakes were filled in the space among the individual curly graphene nanoribbons to avoid the stacking of CGNR. Fig. 2d shows the morphology of the CGNR/PANI/MnO2, and it was observed that the MnO2 nanoneedles were dispersed uniformly over the surfaces of the CGNR and PANI. The MnO2 nanoneedles could further restrain the stacking of the CGNR, but the CGNR also functioned as stable substrates to support the MnO2 nanoneedles to maintain their physical structure and accelerate electron transfer, which could maximize the capacitance of MnO2. | ||
| Fig. 2 SEM images of the (a) MWCNT, (b) CGNR, (c) CGNR/PANI, and (d) CGNR/PANI/MnO2. | ||
Fig. 3a shows TEM images of the CGNR, showing that the CNTs had been unzipped in to CGNR. Fig. 3b presents a TEM image of the CGNR/PANI/MnO2. MnO2 nanoneedles (as pointed by the arrows) with a diameter of ∼10 nm and length of ∼100 nm dispersed uniformly on the surfaces of the CGNR and PANI were clearly observed. The EDS result in Fig. 3c showed that C, N, O, and Mn were presented in the CGNR/PANI/MnO2, which confirmed the existence of PANI and MnO2 in the composite. The amount of manganese dioxide in the nanocomposite was measured by EDS, and the result showed that the weight fraction of Mn was 11.29%, representing 16.01% of MnO2 in the composite, which was the optimal amount of MnO2 in the CGNR/PANI/MnO2 composite (details in the ESI†).
 | ||
| Fig. 3 (a) TEM image of CGNR, and (b) TEM and (c) EDS images of the CGNR/PANI/MnO2 composite. | ||
Typical X-ray diffraction (XRD) patterns of the CGNR, PANI, MnO2 and CGNR/PANI/MnO2 are shown in Fig. 4a. The XRD pattern of the CGNR exhibited a broad peak at 2θ = 25.2° corresponding to the (002), indicating the reduction of the OCGNR and formation of CGNR. The XRD pattern of the PANI showed peaks at 14.9° (011), 20.6° (021) and 25.8° (200) of the emeraldine salt form of PANI. The XRD pattern of MnO2 displayed diffraction peaks at 12.7° (110), 18.1° (200), 28.8° (310), 37.4° (211), 49.8° (411), and 60.2° (521), which were indexed to a pure tetragonal phase of α-MnO2 (JCPDS 44-0141).43 The XRD pattern of the CGNR/PANI/MnO2 showed all the characteristic peaks of CGNR, PANI and MnO2, confirming the successful synthesis of the CGNR/PANI/MnO2 composite.
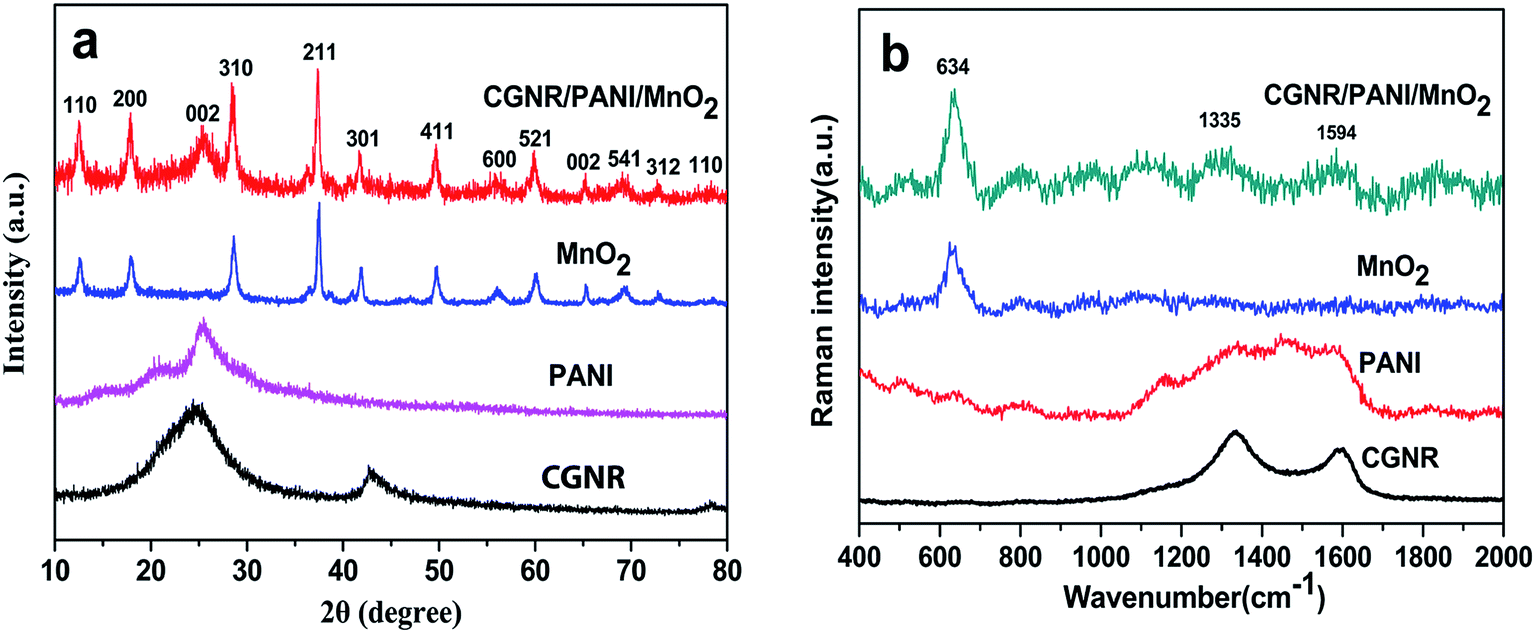 | ||
| Fig. 4 (a) XRD patterns of the CGNR, PANI, MnO2, and CGNR/PANI/MnO2, and (b) Raman spectra of the CGNR, PANI, MnO2, and CGNR/PANI/MnO2. | ||
Fig. 4b presents the Raman spectra of the CGNR, PANI, MnO2, and CGNR/PANI/MnO2. The CGNR displayed two typical peaks at 1335 cm−1 and 1594 cm−1 corresponding to the in-plane bond stretching motion of C sp2 atoms (G band) and the benzenoid rings of CGNR (D-band). The PANI displayed several peaks at 518, 1176, 1329, 1482, and 1592 cm−1, which were attributed to the out-of-plane C–N–C torsion, imine deformation, in-plane ring deformation, C–N+ stretching, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching of quinoid, and C–C stretching of benzenoid.19 MnO2 displayed a sharp peak at 630 cm−1 corresponding to Mn–O lattice vibration in MnO2 nanoneedles. The CGNR/PANI/MnO2 composite displayed all the characteristic peaks mentioned above, further confirming the formation of the ternary nanocomposite.
N stretching of quinoid, and C–C stretching of benzenoid.19 MnO2 displayed a sharp peak at 630 cm−1 corresponding to Mn–O lattice vibration in MnO2 nanoneedles. The CGNR/PANI/MnO2 composite displayed all the characteristic peaks mentioned above, further confirming the formation of the ternary nanocomposite.
The CGNR/PANI/MnO2 composite was also characterized by XPS. Fig. 5a shows the XPS survey scan spectra; Mn (2p1/2, 2p3/2), N 1s, O 1s and C 1s were observed. Fig. 5b shows details from the XPS for the binding energy range of the relevant Mn species. There were two main peaks between 635 and 660 eV that were resolved to 642.1 eV and 653.8 eV for Mn 2p3/2 and Mn 2p1/2, respectively, which were consistent with the well-characterized MnO2 XPS spectra in the literature.27
 | ||
| Fig. 5 (a) XPS survey spectra of the CGNR/PANI/MnO2 composite and (b) the details from the XPS for the binding energy range of the relevant Mn species. | ||
Fig. 6a shows the N2 adsorption/desorption isotherms of CGNR and CGNR/PANI/MnO2, both adsorption/desorption isotherms showed type IV isotherms with a hysteresis loop at a relative pressure (P/P0) of 0.45–1, indicating the mesoporous structure of the two samples.44 This showed that the integration of PANI and MnO2 did not ruin the mesoporous structure of the CGNR, and these mesopores were essentially important to contribute to the capacitance. From the adsorption/desorption isotherms, the BET surface area of CGNR was estimated to be 229 m2 g−1, which was much higher than that of the pristine MWCNTs.45 The BET surface area of the CGNR/PANI/MnO2 was estimated to be 263 m2 g−1, which was larger than that of the CGNR, which may be because PANI and MnO2 promoted the specific surface area through restraining stacking and agglomeration of the CGNR. Fig. 6b shows the pore size distribution curves estimated by Barrett–Joyner–Halenda analysis; the curves showed an intensive pore size distribution of ∼3.8 nm. In addition, the adsorption average pore width of the CGNR/PANI/MnO2 was 5.8 nm, which was smaller than that of the CGNR (6.2 nm) and the pore volume of the CGNR (0.339 cm3 g−1) was smaller than that of the CGNR/PANI/MnO2 (0.356 cm3 g−1), which were beneficial for the ion access.
 | ||
| Fig. 6 (a) N2 adsorption/desorption isotherms of CGNR and CGNR/PANI/MnO2 and (b) pore size distribution curves of the CGNR and CGNR/PANI/MnO2. | ||
Electrochemical performance
The electrochemical performance of the CGNR, PANI, MnO2, and CGNR/PANI/MnO2 was evaluated in 1 M H2SO4 by cyclic voltammetry (CV) and galvanostatic charge–discharge testing. Fig. 7a shows the CV curves of the CGNR, PANI, MnO2, and CGNR/PANI/MnO2 composite at a scan rate of 50 mV s−1. The CV curves of the CGNR, MnO2, and CGNR/PANI/MnO2 all displayed a rectangle shape, indicating good supercapacitive characteristic,46 whereas the CV curve of the PANI displayed two pairs of redox peaks, indicating the redox transition of PANI from a semiconducting state (leucoemeraldine form) to the conducting state (polaronic emeraldine form).32 In particular, CGNR/PANI/MnO2 showed the highest current density, representing the largest specific capacitance. Fig. 7b shows the CV curves of the CGNR/PANI/MnO2 at different scan rates and all the CV curves showed a good rectangular shape, demonstrating good rate performance of the electrode material. | ||
| Fig. 7 (a) Cyclic voltammograms of the CGNR, PANI, MnO2, and CGNR/PANI/MnO2 at a scan rate of 50 mV s−1, and (b) cyclic voltammograms of the CGNR/PANI/MnO2 at different scan rates. | ||
Galvanostatic charge/discharge testing was carried out in 1 M H2SO4. Fig. 8a shows the charge/discharge curves of the CGNR, PANI, MnO2, and CGNR/PANI/MnO2 electrodes at a current density of 1 A g−1. All curves displayed linear responses, showing good agreement with the supercapacitor characteristics.47 The specific capacitance C was calculated using the following equation:48

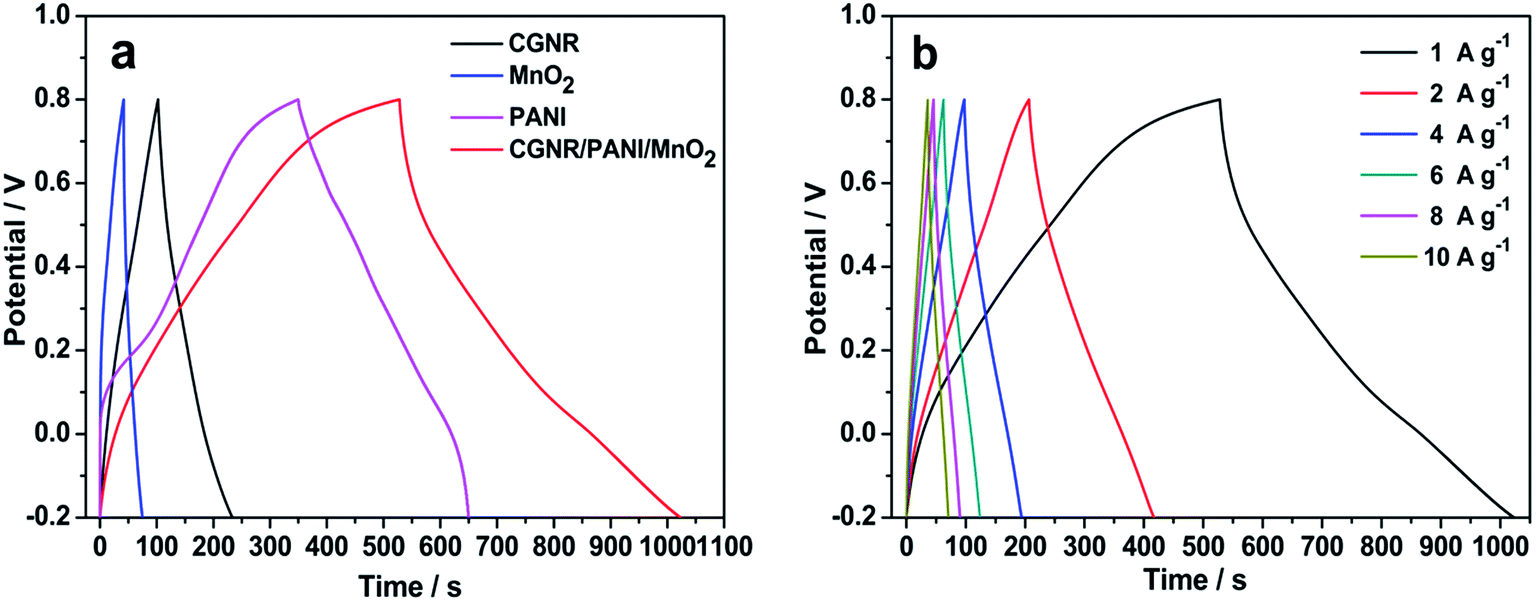 | ||
| Fig. 8 Galvanostatic charge/discharge curves of (a) the CGNR, PANI, MnO2, CGNR/PANI/MnO2 at 1 A g−1 and (b) the CGNR/PANI/MnO2 at different current densities. | ||
Fig. 8b shows the specific capacitance of the CGNR/PANI/MnO2 at different current densities from 1 to 10 A g−1. Fig. 9 shows the specific capacitance of the CGNR/PANI/MnO2 as a function of the current density, and the corresponding data of the CGNR, PANI, MnO2 are also shown for comparison. As shown, the specific capacitance decreased with increasing current density, and the CGNR/PANI/MnO2 electrode delivered the largest capacitance at all the discharge current densities. At a very high current density of 10 A g−1, the CGNR/PANI/MnO2 electrode delivered a remarkable specific capacitance of 348 F g−1, which was among the highest value in the literature.25,41,42
 | ||
| Fig. 9 Specific capacitance of the CGNR, PANI, MnO2 and CGNR/PANI/MnO2 at different current densities from 1 to 10 A g−1. | ||
EIS is an efficient way to measure the electrochemical properties of electrode materials. The impedance spectra were obtained over the frequency range from 105 Hz to 0.01 Hz at the open circuit potential. Fig. 10 shows a Nyquist plot of the CGNR/PANI/MnO2 composite, and there was an approximate semi-circle in the high frequency region, followed by a near-linear section in the low frequency region. In the high frequency region, the equivalent series resistance (ESR, intersection on the real axis at high frequency) and charge transfer resistance (Rct, the diameter of the semicircle) were estimated to be 3.6 Ω and 0.8 Ω, respectively.49 In the low frequency range, a nearly vertical line represented the very good capacitive characteristics of the electrode.50 The low internal resistance demonstrated that the unique structure of the CGNR/PANI/MnO2 electrode facilitated ion and electron transfer during charging and discharging.
 | ||
| Fig. 10 Nyquist plot of the CGNR/PANI/MnO2 electrode. | ||
The electrochemical stability of the electrodes was investigated at a scan rate of 50 mV s−1 for 5000 cycles. Fig. 11 displays the specific capacitance retention as a function of the cycle number. The specific capacitance of PANI and MnO2 decreased sharply over cycling, and only retained 18% and 15% of its initial capacitance after 5000 cycles, whereas the CGNR had nearly no capacitance loss after cycling. The specific capacitance of the CGNR/PANI/MnO2 electrode decreased slightly, retained 81.1% of the initial capacitance after 5000 cycles, demonstrating enhanced cycling stability.
 | ||
| Fig. 11 Cycling stability of the CGNR, PANI, MnO2 and CGNR/PANI/MnO2 electrodes. | ||
Overall, the enhanced capacitive performance was ascribed to the unique structure and the synergic effect of the three components. The CGNR with a porous structure and good electric conductivity was functioned as supports to facilitate ion and electron transfer and to avoid the aggregation of PANI and MnO2. On the other hand, the PANI nanoflakes and MnO2 nanoneedles alleviated the stacking of the CGNR, maintaining a high surface area and mesoporous structure.
The CGNR/PANI/MnO2 supercapacitor was also fabricated and investigated by CV and galvanostatic charge/discharge via a symmetric two-electrode configuration. Fig. 12a displays the CV curves of the symmetric supercapacitor device at scan rates ranging from 5 mV s−1 to 100 mV s−1, and all the CV curves displayed a very good rectangular shape in the voltage window of 1 V, representing remarkable supercapacitor characteristics. Fig. 12b shows the charge/discharge curves at various scan rates, and again linear charge/discharge curves were observed. The specific capacitance of the symmetric supercapacitor was 103 F g−1 (based on the total mass in the supercapacitor) at 0.5 A g−1, which decreased to 81 F g−1 with increasing current density to 5 A g−1.
 | ||
| Fig. 12 (a) CV curves of the CGNR/PANI/MnO2 in a two-electrode configuration at scan rates from 5 to 100 mV s−1 and (b) galvanostatic charge/discharge curves of two-electrode supercapacitor at different current densities. | ||
Energy density and power density are two important performance indicators for supercapacitors. The energy density and power density were estimated using the equations:51

where E is the energy density of the supercapacitor (W h kg−1), P is the power density of the supercapacitor (W kg−1), C is the specific capacitance of the supercapacitor, and t is the discharge time. A Ragone plot using the data in Fig. 12b was constructed, and the maximum energy density was 14.3 W h kg−1.

Fig. 13 shows the Ragone plot, which also contains the comparative recent MnO2 or PANI based supercapacitor data re-plotted from the literature,15,52,53 indicating a much better energy density and power density combination.
 | ||
| Fig. 13 A Ragone plot constructed from data in Fig. 12b, together with some comparative re-plotted data of ternary material-based supercapacitors. | ||
Conclusions
We successfully prepared CGNR/PANI/MnO2 nanocomposites, in which MWCNTs were tailored into CGNR and formed a conductive 3D frame, aniline was polymerized in situ in the CGNR, and MnO2 nanoneedles were interspersed uniformly on the CGNR and PANI to form a three-dimensional architecture. The CGNR/PANI/MnO2 nanocomposite displayed a high BET surface area of 263 m2 g−1 and the morphology and structure were studied carefully. The CGNR/PANI/MnO2 electrode delivered a high specific capacitance of 496 F g−1 at a current density of 1 A g−1, reducing to 348 F g−1 at 10 A g−1, and the capacitance retained 81.1% of its initial value after 5000 cycles. The two-electrode configuration testing showed that the CGNR/PANI/MnO2 had a better energy density and power density combination than some MnO2 or PANI based composites reported in the literature. The enhanced electrochemical performance was attributed to the unique structure of the composite, in which PANI and MnO2 were supported by the 3D CGNR frame with a high surface area and high conductivity, which was beneficial for ion and electron transfer.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 21271069, J1210040, 51238002, J1103312), the Science and Technology Program of Hunan Province (Grant No. 2015JC3049), and the Fundamental Research Funds for the Central Universities.References
- S. Li, J. Niu, Y. C. Zhao, K. P. So, C. Wang, C. A. Wang and J. Li, Nat. Commun., 2015, 6, 7872 CrossRef CAS PubMed.
- Q. Liu, O. Nayfeh, M. H. Nayfeh and S. T. Yau, Nano Energy, 2013, 2, 133–137 CrossRef CAS.
- X. Xiao, W. Zhou, Y. Kim, I. Ryu, M. Gu, C. Wang, G. Liu, Z. Liu and H. Gao, Adv. Funct. Mater., 2015, 25, 1426–1433 CrossRef CAS.
- X. Cao, Y. Shi, W. Shi, G. Lu, X. Huang, Q. Yan, Q. Zhang and H. Zhang, Small, 2011, 7, 3163–3168 CrossRef CAS PubMed.
- Z. Lei, F. Shi and L. Lu, ACS Appl. Mater. Interfaces, 2012, 4, 1058–1064 CAS.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- C. Xiang, M. Li, M. Zhi, A. Manivannan and N. Wu, J. Power Sources, 2013, 226, 65–70 CrossRef CAS.
- Y. Huang, J. Liang and Y. Chen, Small, 2012, 8, 1805–1834 CrossRef CAS PubMed.
- P. Tang, L. Han and L. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 10506–10515 CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- D. Yu, Q. Qian, L. Wei, W. Jiang, K. Goh, J. Wei, J. Zhang and Y. Chen, Chem. Soc. Rev., 2015, 44, 647–662 RSC.
- H. Gao, F. Xiao, C. B. Ching and H. Duan, ACS Appl. Mater. Interfaces, 2012, 4, 2801–2810 CAS.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- Y. Jin, H. Chen, M. Chen, N. Liu and Q. Li, ACS Appl. Mater. Interfaces, 2013, 5, 3408–3416 CAS.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS.
- J. Yan, J. Liu, Z. Fan, T. Wei and L. Zhang, Carbon, 2012, 50, 2179–2188 CrossRef CAS.
- Z. Yang, M. Liu, C. Zhang, W. W. Tjiu, T. Liu and H. Peng, Angew. Chem., Int. Ed., 2013, 52, 3996–3999 CrossRef CAS PubMed.
- S. Grover, S. Goel, V. Sahu, G. Singh and R. K. Sharma, ACS Sustainable Chem. Eng., 2015, 3, 1460–1469 CrossRef CAS.
- A. Burke, J. Power Sources, 2000, 91, 37–50 CrossRef CAS.
- J. W. Lee, A. S. Hall, J. D. Kim and T. E. Mallouk, Chem. Mater., 2012, 24, 1158–1164 CrossRef CAS.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889–4899 CrossRef CAS.
- Z. Gao, F. Wang, J. Chang, D. Wu, X. Wang, X. Wang, F. Xu, S. Gao and K. Jiang, Electrochim. Acta, 2014, 133, 325–334 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2002, 14, 3946–3952 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184–3190 CrossRef CAS.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, J. Phys. Chem. C, 2008, 112, 15075–15083 CAS.
- M. Liu, W. W. Tjiu, J. Pan, C. Zhang, W. Gao and T. Liu, Nanoscale, 2014, 6, 4233–4242 RSC.
- S. Chen, J. Zhu, X. Wu, Q. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS PubMed.
- Y. Hou, Y. Cheng, T. Hobson and J. Liu, Nano Lett., 2010, 10, 2727–2733 CrossRef CAS PubMed.
- Y. F. Li, S. S. Li, D. L. Zhou, A. J. Wang, P. P. Zhang, C. G. Li and J. J. Feng, J. Solid State Electrochem., 2014, 18, 2521–2527 CrossRef CAS.
- J. Li, H. Xie, Y. Li, J. Liu and Z. Li, J. Power Sources, 2011, 196, 10775–10781 CrossRef CAS.
- J. Xu, K. Wang, S. Z. Zu, B. H. Han and Z. Wei, ACS Nano, 2010, 4, 5019–5026 CrossRef CAS PubMed.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- R. B. Rakhi, W. Chen, D. Cha and H. N. Alshareef, J. Mater. Chem., 2011, 21, 16197–16204 RSC.
- M. Liu, Y. E. Miao, C. Zhang, W. W. Tjiu, Z. Yang, H. Peng and T. Liu, Nanoscale, 2013, 5, 7312–7320 RSC.
- L. L. Jiang, X. Lu, C. Xie, G. Wan, H. Zhang and T. Youhong, J. Phys. Chem. C, 2015, 119, 3903–3910 CAS.
- P. Sekar, B. Anothumakkool and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 7661–7669 CAS.
- C. Yuan, L. Su, B. Gao and X. Zhang, Electrochim. Acta, 2008, 53, 7039–7047 CrossRef CAS.
- Y. Zhou, Z. Y. Qin, L. Li, Y. Zhang, Y. L. Wei, L. F. Wang and M. F. Zhu, Electrochim. Acta, 2010, 55, 3904–3908 CrossRef CAS.
- D. Xu, Q. Xu, K. Wang, J. Chen and Z. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 200–209 CAS.
- J. Shen, C. Yang, X. Li and G. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 8467–8476 CAS.
- X. Yu, K. Li, H. Zhou, S. Wei, C. Zhang, X. Li and Y. Kuang, RSC Adv., 2015, 5, 88950–88957 RSC.
- L. Chen, Z. Song, G. Liu, J. Qiu, C. Yu, J. Qin, L. Ma, F. Tian and W. Liu, J. Phys. Chem. Solids, 2013, 74, 360–365 CrossRef CAS.
- W. Ma, S. Chen, S. Yang, W. Chen, Y. Cheng, Y. Guo, S. Peng, S. Ramakrishna and M. Zhu, J. Power Sources, 2016, 306, 481–488 CrossRef CAS.
- Y. Nie, X. Xie, S. Chen, W. Ding, X. Qi, Y. Wang, J. Wang, W. Li, Z. Wei and M. Shao, Electrochim. Acta, 2016, 187, 153–160 CrossRef CAS.
- Y. Sun, W. Zhang, D. Li, L. Gao, C. Hou, Y. Zhang and Y. Liu, Electrochim. Acta, 2015, 178, 823–828 CrossRef CAS.
- Q. Zhang, L. Li, Y. Wang, Y. Chen, F. He, S. Gai and P. Yang, Electrochim. Acta, 2015, 176, 542–547 CrossRef CAS.
- B. Mu, W. Zhang, S. Shao and A. Wang, Phys. Chem. Chem. Phys., 2014, 16, 7872–7880 RSC.
- C. Fu, A. Mahadevegowda and P. S. Grant, J. Mater. Chem. A, 2015, 3, 14245–14253 CAS.
- Z. Wang, D. O. Carlsson, P. Tammela, K. Hua, P. Zhang, L. Nyholm and M. Strømme, ACS Nano, 2015, 9, 7563–7571 CrossRef CAS PubMed.
- D. Zha, P. Xiong and X. Wang, Electrochim. Acta, 2015, 185, 218–228 CrossRef CAS.
- P. Xiong, C. Hu, Y. Fan, W. Zhang, J. Zhu and X. Wang, J. Power Sources, 2014, 266, 384–392 CrossRef CAS.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2013, 7, 174–182 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02777f |
| This journal is © The Royal Society of Chemistry 2016 |