
Structure and electrochemical properties of multilayer graphene prepared by electrochemical exfoliation of graphite in the presence of benzoate ions
Abstract
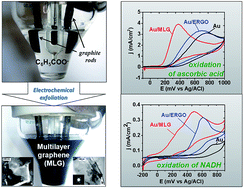
The possibility of obtaining multilayer graphenes–MLGs (11–21 single graphene layers) with slight amounts of oxygen-containing defects (C/O ratio in MLGs ranges from 9 to 25) via the electrochemical exfoliation of graphite in the presence of benzoate anions (MLG1) or benzoate and sulfate anions together (MLG2), under conditions of electrolysis with pulse changing of the electrode polarization potential was established. Composition, morphology, structure and electrochemical properties of MLGs were characterized by CHN-analysis, scanning and transmission electron microscopy, atomic force microscopy, X-ray diffraction, FTIR and UV-Vis spectroscopy, as well as cyclic voltammetry. It was shown that MLG1 and MLG2 can be used directly as electrode materials, since they provide a fast enough heterogeneous electron transfer without any additional reduction (removing of oxygen-containing groups). In particular, the electrocatalytic activity of MLGs in the processes of electrochemical oxidation of such biomarkers as ascorbic acid (MLG1 and MLG2) and NADH (MLG2) exceeds that established for the electrochemical reduced graphene oxide (ERGO), which may be due to the presence of epoxy, aldehyde, carbonyl and peroxide groups in MLGs, unlike ERGO, which can play the role of active sites.
 Please wait while we load your content...
Please wait while we load your content...

