
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel acid-catalyzed rearrangement of 2-substituted-3-(2-nitrophenyl)oxiranes for the synthesis of di- and mono-oxalamides†
Vakhid A.
Mamedov
*ab,
Vera L.
Mamedova
ab,
Gul'nas Z.
Khikmatova
b,
Ekaterina V.
Mironova
a,
Dmitry B.
Krivolapov
a,
Olga B.
Bazanova
a,
Denis V.
Chachkov
b,
Sergey A.
Katsyuba
a,
Il'dar Kh
Rizvanov
a and
Shamil K.
Latypov
a
aA. E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center of the Russian Academy of Sciences, Arbuzov str. 8, 420088 Kazan, Russian Federation. E-mail: mamedov@iopc.ru
bKazan National Research Technological University, Karl Marx str. 68, 420015 Kazan, Russian Federation
First published on 11th March 2016
Abstract
A novel one-pot synthetic approach to N1-(2-carboxyaryl)-N2-(aryl or H)oxalamides from 3-(2-nitroaryl)oxirane-2-carboxamides via the classical Meinwald rearrangement and a new rearrangement sequence has been developed. The methodology is applicable to the synthesis of N-(2-carboxyphenyl)aryloxalmonoamides from (3-(2-nitrophenyl)oxiran-2-yl)(aryl)methanones. The method is operationally simple and high yielding, thus providing a new useful formula for both anthranilic acid derivatives and oxalamides.
Introduction
Oxiranes are one of the most versatile classes of organic compounds available to the synthetic chemist.1 They can be prepared by a wide variety of methods.2 One of the most frequently used atom economical reactions of oxiranes is their rearrangement to carbonyl compounds, and a number of reagents including a variety of Lewis acids3 have been elaborated for this purpose. In principle, for trisubstituted oxiranes two types of rearrangements are possible depending on the migration pathways following the Lewis acid promoted C–O bond cleavage (Scheme 1). The rearrangement of I with hydride (path a) or the alkyl/aryl migration (path b) would lead to ketone II or aldehyde III, respectively.3a,4 The synthetic applications of oxiranes have been the subject of a number of reviews.1a,2,5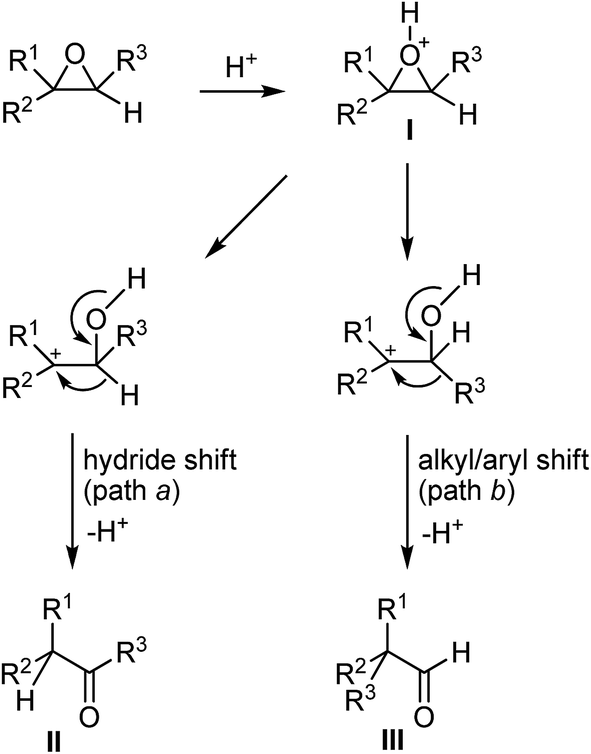 | ||
| Scheme 1 Possible rearrangements of the trisubstituted oxiranes. | ||
The promise of increased chemo-, regio-, and stereoselectivity available via transition metal catalysis6 has led investigators to study the interactions of oxiranes with transition metal complexes, and a number of interesting and useful isomerization reactions have been reported. Notably, oxiranes activated by adjacent aryl, vinyl, silyl, or carbonyl substituents are isomerized to carbonyl compounds or allylic alcohols by complexes of Rh,7 Pd,8 Mo,9 Sm,10 Fe11 and In.12
Unlike all the considered reactions proceeding depending on the structure of oxirane and applied conditions of rearrangement on the path a or b, our strategy included the use of oxiranes, containing substituents with functional groups instead of the usual alkyl or aryl substituents. These functional groups promote an intramolecular condensation (cyclization) of intermediate ketone II or aldehyde III formed as a result of the above two transformations. Recently, our group reported a novel metal-free transannulation reaction of N,3-diaryloxirane-2-carboxamides (AOCAs) involving a one-pot acid-catalyzed Meinwald rearrangement and intramolecular Friedel–Crafts alkylation processes allowing to synthesize various 3-arylquinolin-2(1H)-ones in high yields (Scheme 2).13 This novel approach features not only a metal-free bond formation but also an exclusive 1,2-aryl migration.
 | ||
| Scheme 2 Our previous work and this work. | ||
During our studies on the ring-opening/ring-closure reactions of AOCAs, we attempted to use N-aryl-3-(2-nitroaryl)oxirane-2-carboxamides (obtained from 2-nitrobenzaldehydes and 2-chloro-N-arylacetamides) with the aim of expanding the scope of the reaction. We found that compounds with a newly formed oxalamide chain were obtained instead of expected 3-(2-nitroaryl)quinolin-2(1H)-ones when the reactions were carried out in refluxing AcOH in the presence of H2SO4. As far as we know, there has been no report on the synthesis of unsymmetrical oxalamides via the rearrangement yet. Herein, we report this novel acid-catalyzed rearrangement of AOCAs in AcOH, which proceeds through a cascade of the ring-opening/ring-closure/ring-closure/ring-opening/ring-opening processes.
The salient features of our method are as follows: (1) a variety of aldehydes 1 and chloroacetamides 2 are readily available and the rapid synthesis of 3 with diverse substitution patterns are possible; (2) only two steps are necessary beginning with the starting materials to the products 4; (3) the facile isolation of 3 and 4 are accomplished by a simple aqueous workup.
Results and discussion
The procedure of the Darzens condensation is the same as that described for AOCAs,13 except that only 2-nitrobenzaldehydes instead of variously substituted aromatic aldehydes with chloroacetamides were used and the reactions were carried out at room temperature for 7 h. The mixtures of cis- and trans-isomers of 3-(2-nitroaryl)oxirane-2-carboxamides (3) with the predominance of the trans-isomer were easily purified from the cis-isomer by washing with ether (Table 1).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 1 | R1 | 2 a | R2 | Product | trans/cisb | Yieldtrans,c % |
| a 2-Chloro-N-arylacetamides 2b–f were obtained on a 0.1 mol scale at 0–15 °C by reacting chloroacetylchloride with an equimolar amounts of corresponding aniline and Et3N. The compounds 2a and 2g are commercially available. b Ratio was determined by 1H NMR of the crude products. c Yields refer to isolated trans-isomers of 3. d cis-Isomer of this compound was obtained early.14 | |||||||
| 1 | 1a | H | 2a | Ph | 3a d | 1/0.12 | 87 |
| 2 | 1a | H | 2b | 4-BrC6H4 | 3b | 1/0.14 | 85 |
| 3 | 1a | H | 2c | 3-MeC6H4 | 3c | 1/0.19 | 76 |
| 4 | 1a | H | 2d | 4-MeOC6H4 | 3d | 1/0.29 | 62 |
| 5 | 1a | H | 2e | 3-NO2C6H4 | 3e | 1/0.07 | 91 |
| 6 | 1a | H | 2f | 4-CO2EtC6H4 | 3f | 1/0.09 | 89 |
| 7 | 1b | Cl | 2a | Ph | 3g | 1/0.30 | 61 |
| 8 | 1a | H | 2g | H | 3h | 1/0.00 | 100 |
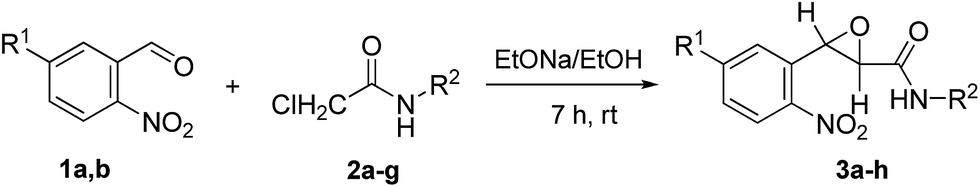
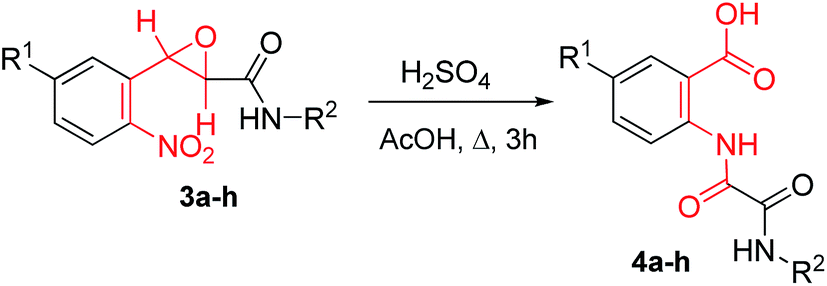
The structures of 3a–h were proved by variety of 1D/2D NMR correlation methods (see ESI†).15 First of all to clarify the optimal reaction conditions we examined the rearrangement of trans-3-(2-nitrophenyl)-N-phenyloxirane-2-carboxamide (3a). After a brief survey of the reaction conditions, we have found that the product 4a is obtained in almost quantitative yield at reflux for 3 h in AcOH with 1 equiv. of H2SO4 (Table 2, entry 1). The reflux of 3a in both MeCN (with 1 equiv. H2SO4) and AcOH for 3 h resulted in the mixtures containing 30 and 10% (determined by 1H NMR) of the desired product 4a, respectively. Further optimization of the reaction conditions was carried out with trans-3-(2-nitrophenyl)oxirane-2-carboxamide (3h). The reflux of 3h in H2O with 1 equiv. H2SO4 for 5 h or its storage at room temp in AcOH with 1 equiv. of H2SO4 for 24 h gave 57 and 75% of the product 4h, respectively. However, an almost quantitative yield of the rearrangement product was achieved when 1 equiv. of H2SO4 was used in boiling AcOH for 3 h (entry 8). The latter condition was used for the rearrangement of all the compounds 3. The rearrangement proceeds equally well with the compounds 3 containing various substituents in an anilide moiety, no matter whether it is a strong electron donating (entry 4) or a strong electron withdrawing (entry 5) group. Interestingly, under the rearrangement conditions the ester group (entry 6) is not subjected to hydrolysis and it can be used in further transformations. The presence of the chlorine atom with the −I and +M electronic effects in the aldehyde component does not influence the yield of the rearrangement product (entry 7).
To our delight 3-(2-nitrophenyl)oxirane-2-carboxamides undergo the rearrangement with the formation of compounds which can be considered both as anthranilic acid and as unsymmetrical oxalamide derivatives. Anthranilic acid derivatives are potential anticancer agents16 and the ligands for farnesoid X receptor.17 Oxalamides also represent a key framework of many bioactive compounds.18 They have been developed as acetylcholine esterase inhibitors,19 C5a inhibitors,20 nitric oxide synthase inhibitors,21 anti-HIV agents,22 antiepileptic drugs,23 HIV integrase inhibitors,24 HIV-1 proteas inhibitors,25 cephalosporin bactericides26 and chemioterapic agents.27 Considering the well documented medicinal utility of anthranilic acid and oxalamide derivatives, these tethered combinations of the two scaffolds afford new opportunities to probe their biological activity.
Based on above results and literature reports,3a,4,28a–c a plausible mechanism for the rearrangement was proposed. First, the process was believed to proceed through the classical Meinwald rearrangement (Scheme 1, path a) of 3-(2-nitrophenyl)oxirane-2-carboxamides with the cleavage of the C2–O bond in its initial stage (3 → A). The resulting ketone A bearing an active α-methylene group undergoes a new rearrangement according to the mechanism of the known Baeyer–Drewson indigo synthesis28a–c with the formation of intermediate C, which is further subjected to acid-catalyzed ring opening (either C → D → E through pathway 1 or C → I → J → F through pathway 2) involving hydration and dehydration processes (E → F → G → H → 4 through pathway 1 and F → G → H → 4 through pathway 2) with the participation of nitro-group and α-methylene functionalities. As a result, the reduction of the nitro-group and the transformation of the C3 atom of epoxide to the carboxylic functionality occurs (Scheme 3).
 | ||
| Scheme 3 Proposed mechanisms of the rearrangement. | ||
The structures of 4a–h were established unambiguously by various 1D/2D NMR correlation methods.15 First, the proton spin systems of the Ar1 and Ar2 moieties were identified by COSY/TOCSY methods. After that, the structures of both halves up to carbonyl groups (C1 and C2) were established (boldfaced on Fig. 1) from the 1H–13C and 1H–15N HSQC/HMBC connectivities. Finally, both halves were linked into a single whole on the basis of the NOEs between protons of these two fragments:
 | ||
| Fig. 1 Structure of 4a with principle NMR correlations (1H–13C/15N HMBCs – black arrow, NOEs – blue arrow). | ||
Structures of the compounds 4a,b were further confirmed by single-crystal X-ray analyses (Fig. 2).
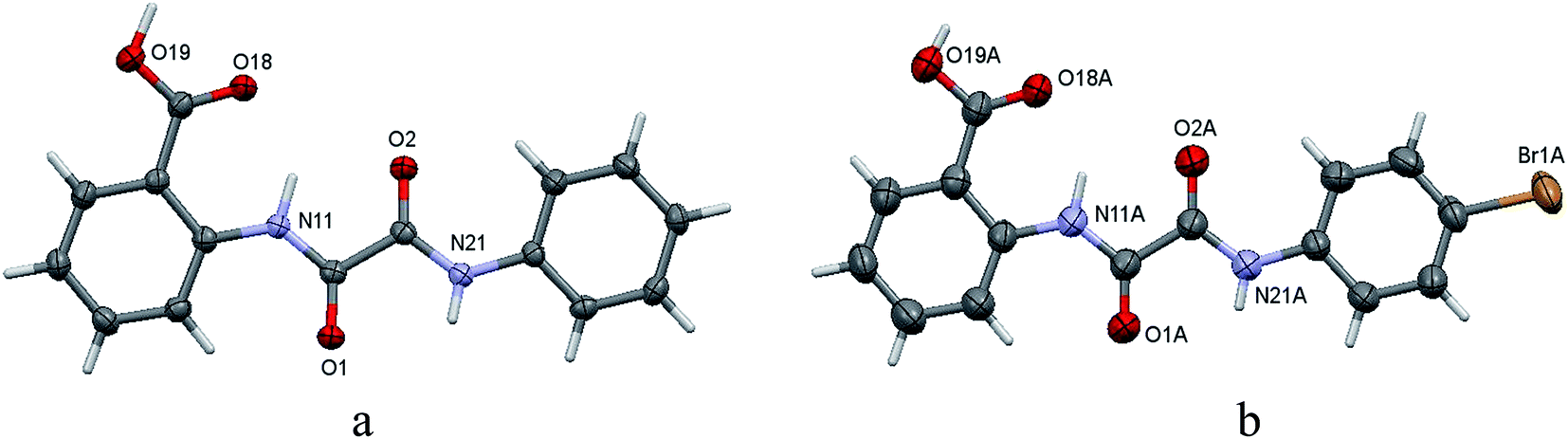 | ||
| Fig. 2 ORTEP plot of compounds 4a (a) and 4b (b) partial numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. H-atoms are represented in stick mode for clarity. | ||
It should be pointed out that a series of synthetic methods for oxalamides have been described in the past decades.29 However, only five examples of the synthetic methods for unsymmetrical oxalamides are known. The first is traditional and based on the condensation of corresponding carboxylic acids with amines, which needs either activating agents or conversion into more reactive derivatives.30 The next three methods include the direct amidation of isocyanates,31 α-keto benzotriazole32 and trichloropyruvamides with amines.33 The fifth method is a novel one and is based on green H2O2-promoted oxidative amidation of 2-oxoaldehydes with amines.34 Nevertheless, these methods have several drawbacks, such as harsh conditions, expensive reagents, poor atom-efficiency and limited substrate scope. Method proposed in this study demonstrated a new, efficient and metal-free synthesis of unsymmetrical oxalamides via novel rearrangement of easily available 3-(2-nitroaryl)-oxirane-2-carboxamides.
With this result in hand, we proceeded with the study of the scope of the rearrangement. As can be seen from the suggested mechanism of the rearrangement (Scheme 3) the transformation of 3-(2-nitroaryl)oxirane-2-carboxamides to the oxalamides involves the 3-(2-nitroaryl)oxirane fragment only. Thus, seeking to expand accessible skeletal diversity using the same reaction conditions, we anticipated that use of (3-(2-nitroaryl)oxiran-2-yl)(aryl)methanones (6a–d) with the same necessary fragment would facilitate access to 2-(2-oxo-2-arylacetamido)benzoic acids (7a–d). Indeed the refluxing of oxiranes 6a–d in AcOH in the presence of catalytic amount of H2SO4 provides the desired mono-oxalamides 7a–d in good yields. Moreover, the reaction of 2-nitrobenzaldehyde (1a) and 5-chloro-2-nitrobenzaldehyde (1b) with 2-chloro-1-(4-tolyl)ethanone (5a), 2-chloro-1-(4-methoxyphenyl)ethanone (5b) and 2-chloro-1-(3-nitrophenyl)ethanone (5c) under the Darzens condensation condition proceeded smoothly, and (3-(2-nitrophenyl)oxiran-2-yl)(aryl)methanones 6a–d were obtained in quantitative yields (Table 3). It should be pointed out that in this case, in contrast to the reactions of chloroacetanilides 2,13 the process proceeds with high stereoselectivity with the formation of only trans-isomers of oxiranes 6a–d as the only products.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | 1 | R1 | 5 a , b | Hal | R2 | Product 6 | Product 7 | Yield,c % |
| a (Chloromethyl)arylketones 5a,b were obtained on a 0.1 mol scale at 10–20 °C by reaction of 1 equiv. chloroacetylchloride with toluene (100 mL) and anisole (100 mL), respectively, with the use of 1.5 equiv. AlCl3. b (Bromomethyl)arylketone 5c was obtained on a 0.1 mol scale at 50–55 °C by reaction of 1 equiv. 1-(3-nitrophenyl)ethanone with 1 equiv. bromine in ethanol (100 mL). c Yields refer to isolated products. | ||||||||
| 1 | 1a | H | 5a | Cl | 4-MeC6H4 | 6a | 7a | 75 |
| 2 | 1a | H | 5b | Cl | 4-MeOC6H4 | 6b | 7b | 81 |
| 3 | 1a | H | 5c | Br | 3-NO2C6H4 | 6c | 7c | 86 |
| 4 | 1b | Cl | 5a | Cl | 4-MeC6H4 | 6d | 7d | 89 |

The structures of all compounds were proved by variety of 1D/2D NMR correlation methods (see ESI†).15
Conclusion
In conclusion, we have discovered a new rearrangement of 3-(2-nitrophenyl)-oxirane-2-carboxamides proceeding in boiling AcOH in the presence of H2SO4. The rearrangement quantitatively produces the N-(2-carboxyaryl)oxalamides as a result of cascade processes involving (a) the classical Meinwald rearrangement in its initial stage with the formation of ketone bearing an active α-methylene group, (b) transformation of carbonyl group of the ketone to the carboxylic functionality, (c) migration of active α-methylene group to the nitrogen atom of already reduced nitro group. The simple reaction conditions offer a potential for employing this method in the synthesis of complex molecules. It is anticipated that this methodology will have versatile applications in the practical syntheses of biologically important pharmaceutical molecules with anthranilic acid and oxalamide moieties. The methodology is applicable to synthesis of N-(2-carboxyphenyl)aryloxalmonoamides from (3-(2-nitrophenyl)oxiran-2-yl)(aryl)methanones. Further extension of the reaction scope and the synthetic applications of this methodology are in progress at our laboratory.Experimental section
General methods
All reagents and solvents were used as purchased, without further purification melting points were determined on a hot-stage apparatus. Infrared (IR) spectra samples in Nujol were recorded on a FT-IR spectrometer Bruker Vector-22 in the 400–4000 cm−1 range at optical resolution of 4 cm−1. The high resolution MALDI mass-spectra were obtained on UltraFlex III TOF/TOF instrument in positive reflectron mode; 2,5-DHB and p-NA were used as matrix and PEG-400 was used for calibration of accurate masses. All NMR experiments were performed with 600, 500 and 400 MHz (600 MHz for 1H NMR; 150.9, 125 and 100.6 MHz for 13C NMR; 60.8 and 50.7 MHz for 15N NMR) spectrometers equipped with 5 mm diameter gradient inverse broad band probehead and a pulsed gradient unit capable of producing magnetic field pulse gradients in the z-direction of 53.5 G cm−1. NMR experiments were carried out at 303 K. DPFGROE35 and TOCSY spectra were obtained using a Hermite-shaped pulse for selective excitation. Chemical shifts (δ in ppm) are referred to the solvent DMSO-d6 (δ = 2.49 ppm for 1H and 39.5 ppm for 13C NMR), to external CD3NO2 (380.2 ppm) for 15N NMR spectra (conversion factor to NH3: −380.2 ppm). 1H–1H coupling constants were computed according to Bally & Rablen's recommendations.36 First the geometry was optimized at the B3LYP/6-31G(d) level. Then NMR single-point calculation of the Fermi contact J values was run at the B3LYP/6-31G(d,p) level. These values were scaled then by a factor of 0.9117. The quantum chemical calculations were performed using a Gaussian 03 software package.37trans-3-(2-Nitrophenyl)-N-phenyloxirane-2-carboxamides (3a).

Tan powder (7.68 g, 0.027 mol, 87% yield): mp 180–181 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.35 (s, 1H, NH), 8.20 (d, J = 8.2 Hz, 1H, H3-Ar1), 7.85 (dd, J = 7.6, 7.6 Hz, 1H, H5-Ar1), 7.65–7.69 (m, 3H, H2,6-Ar2; H4-Ar1), 7.59 (d, J = 7.9 Hz, 1H, H6-Ar1), 7.36 (dd, J = 7.9, 7.9 Hz, 2H, H3,5-Ar2), 7.12 (dd, J = 7.6, 7.2 Hz, 1H, H4-Ar2), 4.68 (d, J = 1.8 Hz, 1H, H3), 3.65 (d, J = 1.8 Hz, 1H, H2); 13C NMR (150.9 MHz, DMSO-d6) δ 164.46 (C1), 147.46 (C2-Ar1), 138.23 (C1-Ar2), 134.66 (C5-Ar1), 132.19 (C1-Ar1), 129.50 (C4-Ar1), 128.78 (C3-Ar2), 126.73 (C6-Ar1), 124.65 (C3-Ar1), 123.91 (C4-Ar2), 119.45 (C2-Ar2), 57.37 (C2), 55.14 (C3); 15N NMR (60.8 MHz, DMSO-d6) δ 371.6 (NO2), 132.3 (NH); IR (nujol): ν 3279, 1674, 1604, 1553, 1525 cm−1; HRMS (MALDI) calcd for C15H12N2O4 [M + Cs]+ 416.9846, found 416.9843; anal. calcd for C15H12N2O4: C, 63.38; H, 4.25; N, 9.85; found: C, 63.42; H, 4.13; N, 9.92.
trans-N-(4-Bromophenyl)-3-(2-nitrophenyl)oxirane-2-carboxamide (3b).
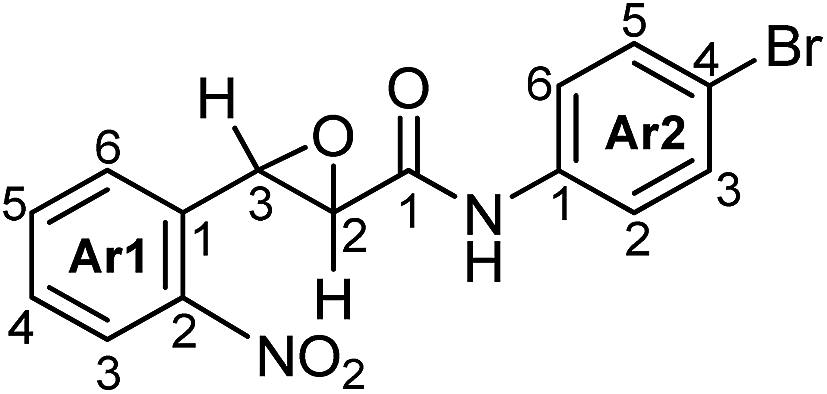
Tan powder (8.33 g, 0.023 mol, 85% yield): mp 191–192 °C; 1H NMR (600 MHz, DMSO-d6): δ 10.50 (s, 1H, NH), 8.19 (dd, J = 8.2, 1.0 Hz, 1H, H3-Ar1), 7.85 (dd, J = 7.7, 7.2 Hz, 1H, H5-Ar1), 7.67 (ddd, J = 8.2, 7.7, 1.0 Hz, 1H, H4-Ar1), 7.64 (d, J = 8.7 Hz, 2H, H2,4-Ar2), 7.58 (d, J = 7.7 Hz, 1H, H6-Ar1), 7.54 (d, J = 8.7 Hz, 2H, H3,5-Ar2), 4.68 (d, J = 1.8 Hz, 1H, H3), 3.64 (d, J = 1.8 Hz, 1H, H2); 13C NMR (100.6 MHz, DMSO-d6) δ 164.74 (C1), 147.45 (C2-Ar1), 137.59 (C1-Ar2), 134.69 (C5-Ar1), 132.09 (C1-Ar1), 131.64 (C4-Ar2), 129.56 (C4-Ar1), 126.73 (C6-Ar1), 124.68 (C3-Ar1), 121.43 (C2-Ar2), 115.61 (C4-Ar2), 57.36 (C2), 55.27 (C3); 15N NMR (60.8 MHz, DMSO-d6) δ 371.5 (NO2), 131.2 (NH). IR (nujol): ν 3362, 1691, 1591, 1521 cm−1; HRMS (MALDI) calcd for C15H11BrN2O4 [M + Cs]+ 494.8951; 496.8932, found 494.8946; 496.8938; anal. calcd for C15H11BrN2O4: C, 49.61; H, 3.05; Br, 22.00; N, 7.71; found: C, 49.84; H, 2.99; Br, 21.84; N, 7.87.
trans-3-(2-Nitrophenyl)-N-3-tolyloxirane-2-carboxamide (3c).

Tan powder (6.12 g, 0.021 mol, 76% yield): mp 140–143 °C; 1H NMR (600 MHz, DMSO-d6): δ 10.29 (s, 1H, NH); 8.21 (dd, J = 8.2, 1.1 Hz, 1H, H3-Ar1), 7.86 (ddd, J = 7.5, 7.2, 1.1 Hz, 1H, H5-Ar1), 7.68 (ddd, J = 7.9, 7.8, 1.4 Hz, 1H, H4-Ar1), 7.59 (d, J = 7.6 Hz, 1H, H2-Ar1), 7.51 (br.s, 1H, H2-Ar2), 7.45 (br.d, J = 8.2 Hz, 1H, H6-Ar2), 7.24 (dd, J = 7.8, 7.7 Hz, 1H, H5-Ar2), 6.94 (d, J = 7.4 Hz, 1H, H4-Ar2), 4.68 (d, J = 2.0 Hz, 1H, H3), 3.65 (d, J = 2.0 Hz, 1H, H2), 2.31 (s, 3H, CH3); 13C NMR (125.8 MHz, DMSO-d6) δ 164.39 (C1), 147.45 (C2-Ar1), 138.17 (C3-Ar2), 138.05 (C1-Ar2), 134.69 (C5-Ar1), 132.24 (C1-Ar1), 129.51 (C4-Ar1), 128.64 (C5-Ar2), 126.73 (C6-Ar1), 124.67 (C4-Ar2), 124.62 (C3-Ar1), 119.96 (C2-Ar2), 116.65 (C6-Ar2), 57.38 (C2), 55.14 (C3), 21.09 (Me); 15N NMR (60.8 MHz, DMSO-d6) δ 371.6 (NO2), 132.5 (NH); IR (nujol): ν 3252, 1667, 1611, 1556, 1524 cm−1; HRMS (MALDI) calcd for C16H14N2O4 [M + Cs]+ 431.0003, found 430.9994; anal. calcd for C16H14N2O4: C, 64.42; H, 4.73; N, 9.39; found: C, 64.86; H, 4.72; N, 9.41.
trans-N-(4-Methoxyphenyl)-3-(2-nitrophenyl)oxirane-2-carboxamide (3d).

Brown powder (5.34 g, 0.017 mol, 62% yield): mp 168–169 °C; 1H NMR (600 MHz, DMSO-d6): δ 10.23 (s, 1H, NH), 8.21 (dd, J = 8.2, 1.1 Hz, 1H, H3-Ar1), 7.86 (ddd, J = 7.6, 7.6, 1.0 Hz, 1H, H5-Ar1), 7.68 (ddd, J = 7.8, 7.8, 1.3 Hz, 1H, H4-Ar1), 7.49–7.60 (m, 3H, H2,4-Ar2; H6-Ar1), 6.93 (d, J = 9.1 Hz, 2H, H3,5-Ar2), 4.68 (d, J = 1.9 Hz, 1H, H3), 3.75 (s, 3H, OCH3), 3.61 (d, J = 1.9 Hz, 1H, H2); 13C NMR (125.8 MHz, DMSO-d6) δ 163.98 (C1), 155.67 (C4-Ar2), 147.47 (C2-Ar1), 134.69 (C5-Ar1), 132.28 (C1-Ar1), 131.40 (C1-Ar2), 129.51 (C4-Ar1), 126.75 (C6-Ar1), 124.68 (C3-Ar1), 121.10 (C2-Ar2), 113.94 (C3-Ar2), 57.44 (C2), 55.17 (OMe), 55.10 (C3); 15N NMR (60.8 MHz, DMSO-d6) δ 371.8 (NO2-Ar1), 130.6 (NH); IR (nujol): ν 3268, 1664, 1608, 1556, 1514 cm−1; HRMS (MALDI) calcd for C16H14N2O5 [M + Na]+ 337.0795, found 337.0814; anal. calcd for C16H14N2O5: C, 61.14; H, 4.50; N, 8.91. Found: C, 61.53; H, 4.48; N, 8.93.
trans-N-(3-Nitrophenyl)-3-(2-nitrophenyl)oxirane-2-carboxamide (3e).

Tan powder (8.09 g, 0.025 mol, 91% yield): mp 177 °C; 1H NMR (600 MHz, DMSO-d6): δ 10.83 (s, 1H, NH), 8.70 (dd, J = 2.2, 2.2 Hz, 1H, H2-Ar2), 8.21 (dd, J = 8.3, 1.0 Hz, 1H, H3-Ar1), 8.01 (dd, J = 8.2, 1.5 Hz, 1H, H6-Ar2), 7.98 (dd, J = 8.2, 1.3 Hz, 1H, H4-Ar2), 7.86 (dd, J = 7.7, 7.3 Hz, 1H, H5-Ar1), 7.68 (dd, J = 7.7, 1.4 Hz, 1H, H4-Ar1), 7.66 (dd, J = 8.2, 8.2 Hz, 1H, H5-Ar2), 7.60 (d, J = 7.7 Hz, 1H, H6-Ar1), 4.73 (d, J = 2.0 Hz, 1H, H3), 3.70 (d, J = 2.0 Hz, 1H, H2); 13C NMR (150.9 MHz, DMSO-d6) δ 165.44 (C1), 147.95 (C3-Ar2), 147.44 (C2-Ar1), 139.27 (C1-Ar2), 134.70 (C5-Ar1), 131.90 (C1-Ar1), 130.29 (C5-Ar2), 129.62 (C4-Ar1), 126.76 (C6-Ar1), 125.50 (C6-Ar2), 124.68 (C3-Ar1), 118.47 (C4-Ar2), 113.72 (C2-Ar2), 57.26 (C2), 55.48 (C3); 15N NMR (60.8 MHz, DMSO-d6) δ 371.4 (NO2-Ar1), 370.3 (NO2-Ar2), 130.7 (NH); IR (nujol): ν 3241, 1673, 1535, 1522 cm−1; HRMS (MALDI) calcd for C15H11N3O6 [M + Na]+ 352.0540, found 352.0559; anal. calcd for C15H11N3O6: C, 54.72; H, 3.37; N, 12.76. Found: C, 54.69; H, 3.28; N, 12.78.
trans-3-(2-Nitrophenyl)-N-(4-ethylcarboxyphenyl)oxirane-2-carboxamide (3f).

White powder (8.56 g, 0.024 mol, 89% yield): mp 180–182 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.86 (br.s, 1H, NH), 8.21 (d, J = 8.2 Hz, 1H, H3-Ar1), 7.96 (d, J = 8.5 Hz, 2H, H3,5-Ar2), 7.86 (dd, J = 7.6, 7.6 Hz, 1H, H5-Ar1), 7.83 (d, J = 8.5 Hz, 2H, H2,6-Ar2), 7.68 (dd, J = 8.0, 7.7 Hz, 1H, H4-Ar1), 7.59 (d, J = 7.2 Hz, 1H, H6-Ar1), 4.71 (d, J = 1.7 Hz, 1H, H3), 3.75 (d, J = 1.7 Hz, 1H, H2), 4.30 (q, J = 7.1 Hz, 2H, OCH2CH3), 1.33 (t, J = 7.1 Hz, 3H, OCH2CH3); 13C NMR (125.8 MHz, DMSO-d6) δ 165.21 (CO2Et), 165.17 (C1), 147.46 (C2-Ar1), 142.57 (C1-Ar2), 134.72 (C5-Ar1), 132.11 (C1-Ar1), 130.24 (C3-Ar2), 129.59 (C4-Ar1), 126.78 (C6-Ar1), 124.93 (C4-Ar2), 124.70 (C3-Ar1), 118.94 (C2-Ar2), 60.46 (OCH2CH3), 57.28 (C2), 55.37 (C3), 14.15 (OCH2CH3); 15N NMR (60.8 MHz, DMSO-d6) δ 371.7 (NO2), 134.1 (NH); IR (nujol): ν 3360, 1711, 1697, 1608, 1595, 1522 cm−1; HRMS (MALDI) calcd for C18H16N2O6 [M + Cs]+ 489.0057, found 489.0055; anal. calcd for C18H16N2O6: C, 60.67; H, 4.53; N, 7.86. Found: C, 60.68; H, 4.48; N, 7.93.
trans-3-(2-Nitro-5-chlorophenyl)-N-phenyloxirane-2-carboxamide (3g).

White powder (5.25 g, 0.016 mol, 61% yield): mp 156–158 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.38 (s, 1H, NH), 8.25 (d, J = 8.8 Hz, 1H, H3-Ar1), 7.75 (dd, J = 8.8, 2.4 Hz, 1H, H4-Ar1), 7.67 (d, J = 8.4 Hz, 2H, H2,6-Ar2), 7.56 (d, J = 2.4 Hz, 1H, H6-Ar1), 7.38 (dd, J = 8.4, 7.5 Hz, 2H, H3,5-Ar2), 7.13 (ddd, J = 7.5, 7.4, 1.0 Hz, 1H, H4-Ar2), 4.72 (d, J = 2.0 Hz, 1H, H3), 3.72 (d, J = 2.0 Hz, 1H, H2); 13C NMR (125.8 MHz, DMSO-d6) δ 164.23 (C1), 146.12 (C2-Ar1), 139.53 (C5-Ar1), 138.19 (C1-Ar2), 134.65 (C1-Ar1), 129.46 (C4-Ar1), 128.82 (C3-Ar2), 126.90 (C3-Ar1), 126.53 (C6-Ar1), 123.99 (C4-Ar2), 119.50 (C2-Ar2), 57.34 (C2), 54.84 (C3); 15N NMR (60.8 MHz, DMSO-d6) δ 368.9 (NO2), 132.6 (NH); IR (nujol): ν 3368, 1693, 1600, 1571, 1533 cm−1; HRMS (MALDI) calcd for C15H11ClN2O4 [M + Na]+ 341.0299, found 341.0308; anal. calcd for C15H11ClN2O4: C, 56.53; H, 3.48; Cl, 11.12; N, 8.79. Found: C, 56.62; H, 3.32; Cl, 11.18; N, 8.92.
trans-3-(2-Nitrophenyl)oxirane-2-carboxamide (3h).

Light yellow powder (5.62 g, 0.027 mol, 100% yield): mp 209–210 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.18 (dd, J = 8.1, 1.0 Hz, 1H, H3-Ar1), 7.82 (ddd, J = 7.8, 7.8, 1.0 Hz, 1H, H5-Ar1), 7.69 (br.s, 1H, NH2); 7.65 (ddd, J = 8.1, 7.8, 1.2 Hz, 1H, H4-Ar1), 7.48 (br.s, 1H, NH2); 7.53 (d, J = 7.7 Hz, 1H, H6-Ar1), 4.52 (d, J = 1.9 Hz, 1H, H3), 3.39 (d, J = 1.9 Hz, 1H, H2); 13C NMR (125.8 MHz, DMSO-d6) δ 168.07 (C1), 147.50 (C2-Ar), 134.68 (C5-Ar), 132.43 (C1-Ar), 129.46 (C4-Ar), 126.73 (C6-Ar), 124.68 (C3-Ar), 57.05 (C2), 54.91 (C3); 15N NMR (50.7 MHz, DMSO-d6) δ 371.8 (NO2), 106.4 (NH2); IR (nujol): ν 3368, 3185, 1665, 1526 cm−1; HRMS (MALDI) calcd for C9H8N2O4 [M + Cs]+ 340.9533, found 340.9536; anal. calcd for C9H8N2O4: C, 51.93; H, 3.87; N, 13.46. Found: C, 52.02; H, 3.85; N, 13.43.
N 1-(2-Carboxyphenyl)-N2-phenyloxalamide (4a).

Light brown powder (0.46 g, 1.62 mmol, 97% yield): mp 227–228 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.75 (s, 1H, N1H), 10.89 (s, 1H, N2H), 8.70 (ddd, J = 8.3, 8.3, 1.0 Hz, 1H, H3-Ar1), 8.08 (dd, J = 8.0, 1.5 Hz, 1H, H6-Ar1), 7.88 (dd, J = 8.7, 1.1 Hz, 2H, H2,6-Ar2), 7.71 (ddd, J = 7.9, 7.9, 1.7 Hz, 1H, H4-Ar1), 7.39 (ddd, J = 8.7, 7.6, 1.1 Hz, 2H, H3,5-Ar2), 7.29 (ddd, J = 7.6, 7.6, 1.1 Hz, 1H, H5-Ar1), 7.18 (ddd, J = 7.4, 7.4, 1.0 Hz, 1H, H4-Ar2); 13C NMR (125.8 MHz, DMSO-d6) δ 168.84 (CO2H), 158.29 (2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 158.03 (1CO), 139.16 (C2-Ar1), 137.41 (C1-Ar2), 134.19 (C4-Ar1), 131.43 (C6-Ar1), 128.65 (C3-Ar2), 124.72 (C4-Ar2), 123.92 (C5-Ar1), 120.60 (C2-Ar2), 119.53 (C3-Ar1), 117.39 (C1-Ar1); 15N NMR (60.8 MHz, DMSO-d6) δ 126.8 (N2), 121.7 (N1); IR (nujol): ν 3329, 3179, 1678, 1586, 1520 cm−1; HRMS (MALDI) calcd for C15H12N2O4 [M + Na]+ 307.0689, found 307.0690; anal. calcd for C15H12N2O4: C, 63.38; H, 4.25; N, 9.85. Found: C, 63.34; H, 4.31; N, 9.93.
O), 158.03 (1CO), 139.16 (C2-Ar1), 137.41 (C1-Ar2), 134.19 (C4-Ar1), 131.43 (C6-Ar1), 128.65 (C3-Ar2), 124.72 (C4-Ar2), 123.92 (C5-Ar1), 120.60 (C2-Ar2), 119.53 (C3-Ar1), 117.39 (C1-Ar1); 15N NMR (60.8 MHz, DMSO-d6) δ 126.8 (N2), 121.7 (N1); IR (nujol): ν 3329, 3179, 1678, 1586, 1520 cm−1; HRMS (MALDI) calcd for C15H12N2O4 [M + Na]+ 307.0689, found 307.0690; anal. calcd for C15H12N2O4: C, 63.38; H, 4.25; N, 9.85. Found: C, 63.34; H, 4.31; N, 9.93.
N 1-(2-Carboxyphenyl)-N2-(4-bromophenyl)oxalamide (4b).
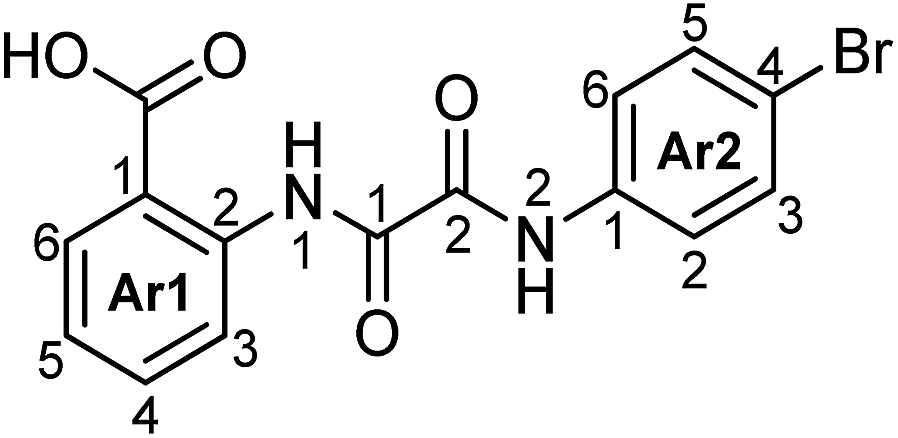
Grey powder (0.61 g, 1.67 mmol, 100% yield): mp 281–283 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.72 (s, 1H, N1H), 11.04 (s, 1H, N2H), 8.68 (d, J = 8.2 Hz, 1H, H3-Ar1), 8.07 (dd, J = 8.2, 1.5 Hz, 1H, H6-Ar1), 7.85 (d, J = 8.7 Hz, 2H, H2,6-Ar2), 7.71 (ddd, J = 7.9, 7.7, 1.5 Hz, 1H, H4-Ar1), 7.57 (d, J = 8.7 Hz, 2H, H3,5-Ar2), 7.28 (ddd, J = 7.7, 7.7, 1.0 Hz, 1H, H5-Ar1); 13C NMR (150.9 MHz, DMSO-d6) δ 168.82 (CO2H), 158.18 (2CO), 158.03 (1CO), 139.10 (C2-Ar1), 136.84 (C1-Ar2), 134.20 (C4-Ar1), 131.49 (C3-Ar2), 131.41 (C6-Ar1), 123.95 (C5-Ar1), 122.54 (C2-Ar2), 119.54 (C3-Ar1), 117.32 (C1-Ar1), 116.65 (C4-Ar2); 15N NMR (60.8 MHz, DMSO-d6) δ 126.6 (N2), 122.2 (N1); IR (nujol): ν 3301, 3189, 1687, 1584, 1519 cm−1; HRMS (MALDI) calcd for C15H11BrN2O4 [M + Na]+ 384.9794; 386.9776, found 384.9795; 386.9786; anal. calcd for C15H11BrN2O4: C, 49.61; H, 3.05; Br, 22.00; N, 7.71. Found: C, 49.38; H, 3.02; Br, 22.07; N, 7.69.
N 1-(2-Carboxyphenyl)-N2-(3-methylphenyl)oxalamide (4c).
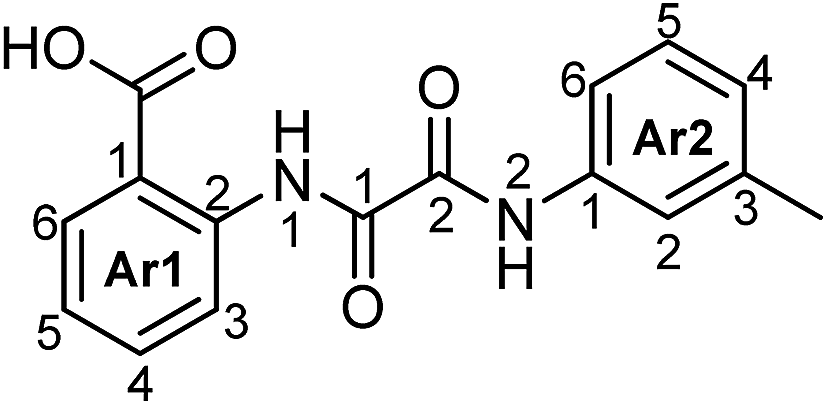
Brown powder (0.46 g, 1.54 mmol, 92% yield): mp 203–205 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.72 (s, 1H, N1H), 10.79 (s, 1H, N2H), 8.70 (d, J = 8.4, 1.0 Hz, 1H, H3-Ar1), 8.08 (dd, J = 7.9, 1.6 Hz, 1H, H6-Ar1), 7.73 (br.s, 1H, H2-Ar2), 7.72 (ddd, J = 7.8, 7.8, 1.7 Hz, 1H, H4-Ar1), 7.64 (br.d, J = 8.3 Hz, 1H, H6-Ar1), 7.29 (ddd, J = 7.9, 7.9, 1.2 Hz, 1H, H5-Ar1), 7.27 (dd, J = 7.8, 7.8 Hz, 1H, H5-Ar2), 7.00 (d, J = 7.6 Hz, 1H, H4-Ar2), 2.33 (s, 3H, CH3); 13C NMR (125.8 MHz, DMSO-d6) δ 168.83 (CO2H), 158.32 (2CO), 157.95 (1CO), 139.16 (C2-Ar1), 137.92 (C3-Ar2), 137.31 (C1-Ar2), 134.24 (C4-Ar1), 131.44 (C6-Ar1), 128.50 (C5-Ar2), 125.44 (C4-Ar2), 123.94 (C5-Ar1), 121.06 (C2-Ar2), 119.53 (C3-Ar1), 117.82 (C6-Ar2), 117.31 (C1-Ar1), 21.12 (Me); 15N NMR (50.7 MHz, DMSO-d6) δ 126.9 (N2), 121.8 (N1); IR ν (nujol): ν 3317, 3187, 1678, 1587, 1528 cm−1; HRMS (MALDI) calcd for C16H14N2O4 [M + Cs]+ 431.0003, found 431.0014; anal. calcd for C16H14N2O4: C, 64.42; H, 4.73; N, 9.39. Found: C, 64.31; H, 4.68; N, 9.42.
N 1-(2-Carboxyphenyl)-N2-(4-methoxyphenyl)oxalamide (4d).

Grey powder (0.50 g, 1.59 mmol, 95% yield): mp 272–273 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.70 (s, 1H, N1H), 10.78 (s, 1H, N2H), 8.69 (ddd, J = 8.4, 7.7, 1.0 Hz, 1H, H3-Ar1), 8.07 (dd, J = 8.2, 1.6 Hz, 1H, H6-Ar1), 7.78 (d, J = 9.1 Hz, 2H, H2,6-Ar2), 7.71 (ddd, J = 7.9, 7.8, 1.6 Hz, 1H, H4-Ar1), 7.28 (ddd, J = 7.8, 7.4, 1.1 Hz, 1H, H5-Ar1), 6.95 (d, J = 9.1 Hz, 2H, H3,5-Ar2), 3.76 (s, 3H, OCH3); 13C NMR (125.8 MHz, DMSO-d6) δ 168.91 (CO2H), 158.56 (2CO), 157.66 (1CO), 156.34 (C4-Ar2), 139.24 (C2-Ar1), 134.31 (C4-Ar1), 131.51 (C6-Ar1), 130.54 (C1-Ar2), 124.01 (C5-Ar1), 122.15 (C2-Ar2), 119.63 (C3-Ar1), 117.37 (C1-Ar1), 113.91 (C3-Ar2), 55.27 (OMe); 15N NMR (50.7 MHz, DMSO-d6) δ 125.6 (N2), 121.8 (N1); IR (nujol): ν 3336, 3218, 1683, 1585, 1525 cm−1; HRMS (MALDI) calcd for C16H14N2O5 [M + Cs]+ 446.9952, found 446.9953; anal. calcd for C16H14N2O5: C, 61.14; H, 4.50; N, 8.91. Found: C, 60.99; H, 4.58; N, 8.97.
N 1-(2-Carboxyphenyl)-N2-(3-nitrophenyl)oxalamide (4e).

Brown powder (0.53, 1.61 mmol, 96% yield): mp 254–256 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.90 (s, 1H, N1H), 11.40 (s, 1H, N2H), 8.90 (dd, J = 2.0, 2.0 Hz, 1H, H2-Ar2), 8.69 (d, J = 8.2 Hz, 1H, H3-Ar1), 8.27 (dd, J = 7.9, 1.2 Hz, 1H, H6-Ar2), 8.08 (dd, J = 7.9, 1.2 Hz, 1H, H6-Ar1), 8.02 (dd, J = 7.9, 1.8 Hz, 1H, H4-Ar2), 7.66–7.72 (m, 2H, H5-Ar2; H4-Ar1), 7.28 (dd, J = 7.8, 7.2 Hz, 1H, H5-Ar1); 13C NMR (150.9 MHz, DMSO-d6) δ 168.93 (CO2H), 158.76 (2CO), 157.68 (1CO), 147.87 (C3-Ar2), 139.05 (C2-Ar1), 138.71 (C1-Ar2), 134.02 (C4-Ar1), 131.43 (C6-Ar1), 130.09 (C5-Ar2), 126.68 (C6-Ar2), 123.97 (C5-Ar1), 119.51 (C3-Ar2), 119.17 (C4-Ar2), 117.80 (C1-Ar1), 114.81 (C2-Ar2); 15N NMR (60.8 MHz, DMSO-d6) δ 370.1 (NO2), 126.6 (N2), 122.2 (N1); IR (nujol): ν 3326, 3188, 1690, 1589, 1532 cm−1; HRMS (MALDI) calcd for C15H11N3O6 [M + Na]+ 352.0540, found 352.0563; anal. calcd for C15H11N3O6: C, 54.72; H, 3.37; N, 12.76. Found: C, 54.82; H, 3.43; N, 12.68.
N 1-(2-Carboxyphenyl)-N2-(4-ethylcarboxypheny)oxalamide (4f).

Light green powder (0.58 g, 1.63 mmol, 97% yield): mp 268–269 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.78 (s, 1H, N1H), 11.18 (s, 1H, N2H), 8.68 (d, J = 8.8 Hz, 1H, H3-Ar1), 8.08 (d, J = 7.7 Hz, 1H, H6-Ar1), 8.04 (d, J = 8.8 Hz, 2H, H2,6-Ar2), 7.96 (d, J = 8.8 Hz, 2H, H3,5-Ar2), 7.70 (dd, J = 7.7, 7.7 Hz, 1H, H4-Ar1), 7.28 (dd, J = 7.7, 7.7 Hz, 1H, H5-Ar1), 4.30 (q, J = 7.1 Hz, 2H, OCH2CH3), 1.32 (t, J = 7.1 Hz, 3H, OCH2CH3); 13C NMR (150.9 MHz, DMSO-d6) δ 168.90 (CO2H), 165.18 (CO2Et), 158.52 (2CO), 157.88 (1CO), 141.74 (C1-Ar2), 139.12 (C2-Ar1), 134.17 (C4-Ar1), 131.45 (C6-Ar1), 129.94 (C3-Ar2), 125.70 (C4-Ar2), 123.97 (C5-Ar1), 120.12 (C2-Ar2), 119.53 (C3-Ar1), 117.47 (C1-Ar1), 60.52 (OCH2CH3), 14.13 (OCH2CH3); 15N NMR (60.8 MHz, DMSO-d6) δ 127.6 (N2), 122.0 (N1); IR (nujol): ν 3355, 3260, 1708, 1688, 1602, 1586, 1524, 1271, 761 cm−1; HRMS (MALDI) calcd for C18H16N2O6 [M + 2Cs − H]+ 620.9034, found 620.9042; anal. calcd for C18H16N2O6: C, 60.67; H, 4.53; N, 7.86. Found: C, 60.59; H, 4.48; N, 7.97.
N 1-(4-Chloro-2-carboxyphenyl)-N2-phenyloxalamide (4g).

Brown powder (0.52 g, 1.63 mmol, 98% yield): mp 242–243 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.84 (s, 1H, N1H), 10.89 (s, 1H, N2H), 8.70 (d, J = 8.9 Hz, 1H, H3-Ar1), 8.02 (d, J = 2.6 Hz, 1H, H6-Ar1), 7.86 (d, J = 7.8 Hz, 2H, H2,6-Ar2), 7.76 (dd, J = 8.9, 2.6 Hz, 1H, H4-Ar1), 7.39 (dd, J = 8.1, 7.5 Hz, 2H, H3,5-Ar2), 7.18 (dd, J = 7.5, 7.4 Hz, 1H, H4-Ar2); 13C NMR (100.9 MHz, DMSO-d6) δ 167.59 (CO2H), 158.34 (2CO), 157.86 (1CO), 137.98 (C2-Ar1), 137.37 (C1-Ar2), 133.58 (C4-Ar1), 130.61 (C6-Ar1), 128.64 (C3-Ar2), 127.51 (C5-Ar1), 124.74 (C4-Ar2), 121.29 (C3-Ar1), 120.61 (C2-Ar2), 119.71 (C1-Ar1); 15N NMR (50.7 MHz, DMSO-d6) δ 127.0 (N2), 121.4 (N1); IR (nujol): ν 3331, 3166, 1679, 1579, 1505 cm−1; HRMS (MALDI) calcd for C15H11ClN2O4 [M + Na]+ 341.0300, found 341.0328; anal. calcd for C15H11ClN2O4: C, 56.53; H, 3.48; Cl, 11.12; N, 8.79. Found: C, 56.59; H, 3.52; Cl, 11.07; N, 8.99.
N-(2-Carboxyphenyl)oxalamide (4h).

Violet powder (0.34 g, 1.62 mmol, 97% yield): mp 267–268 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.51 (s, 1H, N1H), 8.65 (d, J = 8.2 Hz, 1H, H3-Ar), 8.36 (br.s, 1H, NH2), 8.05 (br.s, 1H, NH2), 8.04 (dd, J = 8.0, 1.6 Hz, 1H, H6-Ar), 7.68 (ddd, J = 7.9, 7.9, 1.5 Hz, 1H, H4-Ar), 7.25 (ddd, J = 7.6, 7.6, 1.0 Hz, 1H, H5-Ar); 13C NMR (125.8 MHz, DMSO-d6) δ 168.66 (CO2H), 161.57 (1CO), 158.73 (2CO), 139.19 (C2-Ar), 134.13 (C4-Ar), 131.38 (C6-Ar), 123.70 (C5-Ar), 119.47 (C3-Ar), 117.22 (C1-Ar); 15N NMR (50.7 MHz, DMSO-d6) δ 103.5 (N2), 121.3 (N1); IR (nujol): ν 3538, 3465, 3321, 3168, 1729, 1683, 1592, 1537, 1272, 752 cm−1; HRMS (MALDI) calcd for C9H8N2O4 [M + 2Cs − H]+ 472.8509, found 472.8491; anal. calcd for C9H8N2O4: C, 51.93; H, 3.87; N, 13.46. Found: C, 51.90; H, 3.89; N, 13.42.
trans-(3-(2-Nitrophenyl)oxiran-2-yl)(4-tolyl)methanone (6a).

White powder (7.65 g, 0.027 mol, 100% yield): mp 154 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.21 (dd, J = 8.1, 1.0 Hz, 1H, H3-Ar1), 8.0 (d, J = 8.2 Hz, 2H, H3,5-Ar2), 7.88 (ddd, J = 7.6, 7.5, 0.8 Hz, 1H, H5-Ar1), 7.65–7.71 (m, 2H, H4,6-Ar1), 7.37 (d, J = 8.2 Hz, 2H, H2,6-Ar1), 4.74 (d, J = 2.1 Hz, 1H, H2), 4.56 (d, J = 2.1 Hz, 1H, H3), 2.41 (s, 3H, Me); 13C NMR (100.6 MHz, DMSO-d6) δ 192.14 (1CO), 147.47 (C2-Ar1), 144.72 (C4-Ar2), 134.60 (C5-Ar1), 132.70 (C1-Ar2), 132.42 (C1-Ar1), 129.47 (C4-Ar1), 129.38 (C2-Ar2), 128.50 (C3-Ar2), 127.02 (C6-Ar1), 124.62 (C3-Ar1), 58.34 (C2), 57.15 (C3), 21.19 (Me). IR (nujol): ν 1682, 1522 cm−1; HRMS (MALDI) calcd for C16H13NO4 [M + Cs]+ 415.9894, found 415.9895; anal. calcd for C16H13NO4: C, 67.84; H, 4.63; N, 4.94; found: C, 67.89; H, 4.62; N, 4.98.
trans-(4-Methoxyphenyl)(3-(2-nitrophenyl)oxiran-2-yl)methanone (6b).

White powder (8.08 g, 0.027 mol, 100% yield): mp 141 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 8.2 Hz, 1H, H3-Ar1), 8.09 (d, J = 8.1 Hz, 2H, H2,6-Ar2), 7.87 (dd, J = 7.6, 7.4 Hz, 1H, H5-Ar1), 7.65–7.71 (m, 2H, H6,4-Ar1), 7.08 (d, J = 8.1 Hz, 2H, H3,5-Ar2), 4.71 (d, J = 1.7 Hz, 1H, H2), 4.56 (d, J = 1.7 Hz, 1H, H3), 3.87 (s, 3H, OMe); 13C NMR (125.8 MHz, DMSO-d6) δ 190.82 (1CO), 163.87 (C4-Ar2), 147.45 (C2-Ar1), 134.60 (C5-Ar1), 132.53 (C1-Ar1), 130.87 (C2-Ar2), 129.43 (C4-Ar1), 128.20 (C1-Ar2), 127.05 (C6-Ar1), 124.61 (C3-Ar1), 114.11 (C3-Ar2), 58.17 (C2), 57.03 (C3), 55.58 (OMe). IR (nujol): ν 1678, 1597, 1513; HRMS (MALDI) calcd for C16H13NO5 [M + Cs]+ 431.9843, found 431.9813; anal. calcd for C16H13NO5: C, 64.21; H, 4.38; N, 4.68; found: C, 64.39; H, 4.51; N, 4.77.
trans-(3-Nitrophenyl)(3-(2-nitrophenyl)oxiran-2-yl)methanone (6c).

Grey powder (8.49 g, 0.027 mol, 100% yield): mp 156–157 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.79 (s, 1H, H2-Ar2), 8.53 (d, J = 8.2 Hz, 2H, H4,6-Ar2), 8.23 (d, J = 8.0 Hz, 1H, H3-Ar1), 7.86–7.11 (m, 2H, H5-Ar1, H5-Ar2), 7.67–7.72 (m, 2H, H4,6-Ar1), 4.90 (d, J = 1.9 Hz, 1H, H2), 4.65 (d, J = 1.9 Hz, 1H, H3); 13C NMR (125.8 MHz, DMSO-d6) δ 191.77 (1CO), 148.05 (C3-Ar2), 147.45 (C2-Ar1), 136.05 (C1-Ar2), 134.70 (C5-Ar1), 134.63 (C6-Ar2), 132.15 (C1-Ar1), 130.72 (C5-Ar2), 129.64 (C4-Ar1), 128.17 (C4-Ar2), 127.07 (C6-Ar1), 124.68 (C3-Ar1), 122.71 (C2-Ar2), 58.71 (C2), 57.73 (C3). IR (nujol): ν 1697, 1522 cm−1; HRMS (MALDI) calcd for C15H10N2O6 [M + Cs]+ 446.9588, found 446.9581; anal. calcd for C15H10N2O6: C, 57.28; H, 3.32; N, 8.78; found: C, 57.33; H, 3.21; N, 8.91.
trans-(3-(5-Chloro-2-nitrophenyl)oxiran-2-yl)(4-tolyl)methanone (6d).

Tan powder (8.58 g, 0.027 mol, 100% yield): mp 139 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.24 (d, J = 8.8 Hz, 1H, H3-Ar1), 8.01 (d, J = 8.1 Hz, 2H, H2,6-Ar2), 7.75 (dd, J = 8.8, 2.4 Hz, 1H, H4-Ar1), 7.59 (d, J = 2.4 Hz, 1H, H6-Ar1), 7.37 (d, J = 8.1 Hz, 2H, H3,5-Ar2), 4.81 (d, J = 2.1 Hz, 1H, H2), 4.60 (d, J = 2.1 Hz, 1H, H3), 2.41 (s, 3H, Me); 13C NMR (125.8 MHz, DMSO-d6) δ 191.86 (1CO), 146.17 (C2-Ar1), 144.85 (C4-Ar2), 139.44 (C5-Ar1), 134.86 (C1-Ar1), 132.64 (C1-Ar2), 129.41 (C3-Ar2), 129.35 (C4-Ar1), 128.56 (C2-Ar2), 126.82 (C3-Ar1), 126.71 (C6-Ar1), 58.24 (C2), 56.65 (C3), 21.20 (Me). IR (nujol): ν 1681, 1607, 1516; HRMS (MALDI) calcd for C16H12ClNO4 [M + Cs]+ 449.9504, found 449.9522; anal. calcd for C16H12ClNO4: C, 60.48; H, 3.81; Cl, 11.16; N, 4.41; found: C, 60.31; H, 3.71; N, 4.61.
2-[2-Oxo-2-(4-tolyl)acetamido]benzoic acid (7a).

Brown powder (0.35 g, 1.25 mmol, 75% yield): mp 199–201 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.72 (s, 1H, NH), 8.64 (d, J = 7.5 Hz, 1H, H3-Ar1), 8.14 (d, J = 7.8 Hz, 2H, H2,6-Ar2), 8.04 (dd, J = 8.1, 1.0 Hz, 1H, H6-Ar1), 7.71 (dd, J = 7.6, 7.5 Hz, 1H, H4-Ar1), 7.37 (d, J = 7.8 Hz, 2H, H3,5-Ar2), 7.28 (dd, J = 7.5, 7.9 Hz, 1H, H5-Ar1), 2.42 (s, 3H, Me); 13C NMR (125.8 MHz, DMSO-d6) δ 187.00 (2CO), 169.15 (CO2H), 160.57 (1CO), 145.42 (C4-Ar2), 139.28 (C2-Ar1), 134.19 (C4-Ar1), 131.41 (C6-Ar1), 130.94 (C2-Ar2), 130.38 (C1-Ar2), 129.27 (C3-Ar2), 124.00 (C5-Ar1), 120.13 (C3-Ar1), 117.75 (C1-Ar1), 21.39 (Me). IR (nujol): ν 3363, 1678, 1604, 1586, 1521, 1262 cm−1; HRMS (MALDI) calcd for C16H13NO4 [M + Cs]+ 415.9894, found 415.9887; anal. calcd for C16H13NO4: C, 67.84; H, 4.63; N, 4.94. Found: C, 67.92; H, 4.68; N, 4.71.
2-[2-(4-Methoxyphenyl)-2-oxoacetamido]benzoic acid (7b).

Brown powder (0.40 g, 1.35 mmol, 81% yield): mp 178–181 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H, NH), 8.65 (d, J = 8.3 Hz, 1H, H3-Ar1), 8.27 (d, J = 8.9 Hz, 2H, H2,6-Ar2), 8.05 (d, J = 8.0 Hz, 1H, H6-Ar1), 7.67 (dd, J = 7.2, 8.3 Hz, 1H, H4-Ar1), 7.26 (dd, J = 7.2, 7.8 Hz, 1H, H5-Ar1), 8.10 (d, J = 8.9 Hz, 2H, H3,5-Ar2), 3.88 (s, 3H, OMe); 13C NMR (125.8 MHz, DMSO-d6) δ 185.42 (1CO), 169.12 (CO2H), 164.39 (C4-Ar2), 160.81 (2CO), 139.35 (C2-Ar1), 134.14 (C4-Ar1), 133.47 (C2-Ar2), 131.39 (C6-Ar1), 125.61 (C4-Ar2), 123.88 (C5-Ar1), 120.04 (C3-Ar1), 117.60 (C1-Ar1), 114.10 (C3-Ar2), 55.70 (OMe). IR (nujol): ν 3472, 3258, 1702, 1674, 1601, 1584, 1528, 1260 cm−1; HRMS (MALDI) calcd for C16H13NO5 [M + Cs]+ 431.9843, found 431.9828; anal. calcd for C16H13NO5: C, 64.21; H, 4.38; N, 4.68. Found: C, 64.32; H, 4.37; N, 4.75.
2-[2-(3-Nitrophenyl)-2-oxoacetamido]benzoic acid (7c).

Brown powder (0.45 g, 1.44 mmol, 86% yield): mp 251–252 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.60 (s, 1H, NH), 9.06 (s, 1H, H2-Ar2), 8.70 (d, J = 8.1 Hz, 1H, H3-Ar1), 8.58 (d, J = 7.7 Hz, 1H, H6-Ar2), 8.54 (dd, J = 8.2, 1.5 Hz, 1H, H4-Ar2), 8.09 (dd, J = 7.9, 1.5 Hz, 1H, H6-Ar1), 7.89 (dd, J = 7.9, 8.0 Hz, 1H, H5-Ar2), 7.72 (ddd, J = 7.9, 7.9, 1.5 Hz, 1H, H4-Ar1), 7.30 (dd, J = 7.9, 7.9 Hz, 1H, H5-Ar1); 13C NMR (125.8 MHz, DMSO-d6) δ 185.04 (1CO), 169.14 (CO2H), 159.14 (2CO), 147.38 (C3-Ar2), 139.26 (C2-Ar1), 136.71 (C6-Ar2), 134.46 (C1-Ar2), 134.22 (C4-Ar1), 131.44 (C6-Ar1), 130.14 (C5-Ar2), 128.038 (C4-Ar2), 125.58 (C2-Ar2), 123.99 (C5-Ar1), 119.88 (C3-Ar1), 117.58 (C1-Ar1). IR (nujol): ν 3413, 3189, 1695, 1679, 1588, 1532, 1288 cm−1; HRMS (MALDI) calcd for C15H10N2O6 [M + 2Cs − H]+ 578.8564, found 578.8545; anal. calcd for C15H10N2O6: C, 57.28; H, 3.32; N, 8.78; found: C, 57.34; H, 3.37; N, 8.89.
5-Chloro-2-[2-oxo-2-(4-tolyl)acetamido]benzoic acid (7d).

Light yellow powder (0.47 g, 1.49 mmol, 89% yield): mp 257–258 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H, NH), 8.64 (d, J = 8.9 Hz, 1H, H3-Ar1), 8.13 (d, J = 7.9 Hz, 2H, H2,6-Ar2), 7.97 (br.s, 1H, H6-Ar1), 7.74 (dd, J = 8.8, 1.8 Hz, 1H, H4-Ar1), 7.38 (d, J = 7.9, Hz, 2H, H3,5-Ar2), 2.41 (s, 3H, Me); 13C NMR (125.8 MHz, DMSO-d6) δ 186.46 (1CO), 167.80 (CO2H), 160.35 (2CO), 145.35 (C4-Ar2), 138.03 (C2-Ar1), 133.70 (C4-Ar1), 130.90 (C2-Ar2), 130.51 (C6-Ar1), 130.25 (C1-Ar2), 129.16 (C3-Ar2), 127.55 (C5-Ar1), 121.83 (C3-Ar1), 119.53 (C1-Ar1), 21.32 (Me). IR (nujol): ν 3219, 1703, 1681, 1598, 1577, 1521, 1248 cm−1; HRMS (MALDI) calcd for C16H12ClNO4 [M + 2Cs − H]+ 581.8480, found 581.8455; anal. calcd for C16H12ClNO4: C, 60.48; H, 3.81; Cl, 11.16; N, 4.41; found: C, 60.52; H, 3.73; N, 4.58.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work was partially supported by the Russian scientific foundation (grants No. 14-23-00073 and No. 14-50-00014).References
- (a) J. G. Smith, Synthesis, 1984, 629 CrossRef CAS; (b) E. G. Lewars, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. W. Rees, Pergamon, Oxford, 1984, vol. 7, p. 95 Search PubMed; (c) M. Bartok and K. L. Lang, in The Chemistry of Heterocyclic Compounds: Small Ring Heterocycles, Part 3, ed. A. Hassner, Wiley Intersience, New York, 1983, vol. 42, p. 1 Search PubMed.
- A. S. Rao, S. K. Paknikar and J. G. Kirtane, Tetrahedron, 1983, 39, 2323 CrossRef CAS.
- (a) B. Rickborn, in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon, Oxford, 1991, vol. 3, p. 733 Search PubMed; (b) K. Maruoka, N. Murase, R. Bureau, T. Ooi and H. Yamamoto, Tetrahedron, 1994, 50, 3663 CrossRef CAS; (c) H. O. House, J. Am. Chem. Soc., 1955, 77, 3070 CrossRef CAS.
- R. E. Parker and N. S. Isaacs, Chem. Rev., 1959, 59, 737 CrossRef CAS.
- B. E. Rossiter, in Asymmetric Synthesis, ed. J. D. Morrison, Academic Press, Orlando, FL, 1983, vol. 5, p. 194 Search PubMed.
- L. S. Hegedus, Transition Metals in the Synthesis of Complex Organic Molecules, University Science Books, Menlo Park, CA, 1994 Search PubMed.
- (a) G. Adames, C. Bibby and R. Grigg, J. Chem. Soc., Chem. Commun., 1972, 491 RSC; (b) D. Milstein, O. Buchman and J. Blum, J. Org. Chem., 1977, 42, 2299 CrossRef CAS; (c) D. Milstein, J. Am. Chem. Soc., 1982, 104, 5227 CrossRef CAS.
- (a) M. Suzuki, Y. Oda and R. Noyori, J. Am. Chem. Soc., 1979, 101, 1623 CrossRef CAS; (b) M. Suzuki, A. Watanabe and R. Noyori, J. Am. Chem. Soc., 1980, 102, 2095 CrossRef CAS; (c) T. Hirao, N. Yamada, Y. Ohshiro and T. Agawa, Chem. Lett., 1982, 1997 CrossRef CAS; (d) Y. D. Vankar, N. C. Chaudhuri and S. P. Singh, Synth. Commun., 1986, 16, 1621 CrossRef CAS; (e) G. Visentin, O. Piccolo and G. Consiglio, J. Mol. Catal., 1990, 61, L1 CrossRef CAS; (f) S. Kulasegaram and R. J. Kulawiec, J. Org. Chem., 1997, 62, 6547 CrossRef CAS.
- H. Alper, D. Roches, T. Durst and R. Legault, J. Org. Chem., 1976, 41, 3611 CrossRef CAS.
- J. Prandi, J. L. Namy, G. Menoret and H. B. Kagan, J. Organomet. Chem., 1985, 285, 449 CrossRef CAS.
- (a) R. Aumann, K. Frohlich and H. Ring, Angew. Chem., Int. Ed., 1974, 13, 275 CrossRef; (b) K. Hayakawa and H. Schmid, Helv. Chim. Acta, 1977, 60, 1942 CrossRef CAS; (c) T. Kauffmann, C. Neiteler and G. Neiteler, Chem. Ber., 1994, 127, 659 CrossRef CAS.
- B. C. Ranu and U. Jana, J. Org. Chem., 1998, 63, 8212 CrossRef CAS.
- V. A. Mamedov, V. L. Mamedova, S. F. Kadyrova, G. Z. Khikmatova, A. T. Gubaidullin, I. K. Rizvanov and S. K. Latypov, Tetrahedron, 2015, 71, 2670 CrossRef CAS.
- W.-J. Liu, B.-D. Lv and L.-Z. Gong, Angew. Chem., Int. Ed., 2009, 48, 6503 CrossRef CAS PubMed.
- (a) A. E. Derome, Modern NMR Techniques for Chemistry Research, Pergamon, Cambridge, UK, 1988 Search PubMed; (b) T. I. Atta-ur-Rahman, One and Two Dimensional NMR Spectroscopy, Elsevier, Amsterdam, 1989, p. 578 Search PubMed.
- L. Shi, R. Hu, Y. Wei, Y. Liang, Z. Yang and S. Ke, Eur. J. Med. Chem., 2012, 54, 549 CrossRef CAS PubMed.
- D. Merk, M. Gabler, R. Carrasco Gomez, D. Flesch, T. Hanke, A. Kaiser, C. Lamers, O. Werz, G. Schneider and M. Schubert-Zsilavecz, Bioorg. Med. Chem., 2014, 22, 2447 CrossRef CAS PubMed.
- (a) A. Z. Bialvaei and H. S. Kafil, Curr. Med. Res. Opin., 2015, 31, 707 CrossRef CAS PubMed; (b) A. Kristiansen, M. Grgic, B. Altermark and I. Leiros, J. Antimicrob. Chemother., 2015, 70, 766 CrossRef CAS PubMed; (c) F. J. Barreyro, S. Holod, P. V. Finocchietto, A. M. Camino, J. B. Aquino, A. Avagnina, M. C. Carreras, J. J. Poderoso and G. Gores, Liver Int., 2015, 35, 953 CrossRef CAS PubMed; (d) P. K. Sahoo and P. Behera, Eur. J. Med. Chem., 2010, 45, 909 CrossRef CAS PubMed; (e) F. D. Settimo, J. Med. Chem., 2001, 44, 4359 CrossRef PubMed; (f) A. Michel, P. Downey, J. M. Nicolas and D. Scheller, PLoS One, 2014, 9, e114086 Search PubMed; (g) G. Barta-Szalai, I. Borza and E. Bozo, Bioorg. Med. Chem. Lett., 2004, 14, 3953 CrossRef CAS PubMed.
- K. O. Yerdelen, Med. Chem. Res., 2015, 24, 588 CrossRef CAS.
- T. J. Lanza, P. L. Durette, T. Rollins, S. Siciliano, D. N. Cianciarulo, S. V. Kobayashi, C. G. Caldwell, M. S. Springer and W. K. Hagmann, J. Med. Chem., 1992, 35, 252 CrossRef CAS PubMed.
- H. J. Zhong, L. J. Liu and C. M. Chong, PLoS One, 2014, 9, e92905 Search PubMed.
- (a) H. Otsuki, T. Hishiki, T. Miura, C. Hashimoto, T. Narumi, H. Tamamura, K. Yoshimura, S. Matsushita and T. J. Igarashi, J. Gen. Virol., 2013, 94, 2710 CrossRef CAS PubMed; (b) C. Hashimoto, T. Narumi and H. Otsuki, Bioorg. Med. Chem., 2013, 21, 7884 CrossRef CAS PubMed; (c) T. Narumi, H. Arai, K. Yoshimura, S. Harada, W. Nomura, S. Matsushita and H. Tamamura, Bioorg. Med. Chem., 2011, 19, 6735 CrossRef CAS PubMed.
- A. P. Nikalje, M. Ghodke and A. Girbane, Arch. Pharmacol., 2012, 345, 57 CrossRef CAS PubMed.
- (a) S. B. Singh, F. Pelaez, D. J. Hazuda and R. B. Lingham, Drugs Future, 2005, 30, 277 CrossRef CAS; (b) J. P. Guare, J. S. Wai, R. P. Gomez, N. J. Anthony, S. M. Jolly, A. R. Cortes, J. P. Vacca, P. J. Felock, K. A. Stillmock, W. A. Schleif, G. Moyer, L. J. Gabryelski, L. Jin, I. W. Chen, D. J. Hazuda and S. D. Young, Bioorg. Med. Chem. Lett., 2006, 16, 2900 CrossRef CAS PubMed; (c) T. W. North, A. Villalobos and S. J. Hurwitz, Antimicrob. Agents Chemother., 2014, 58, 3927 CrossRef PubMed.
- (a) P. K. Jadhav and H. W. Man, Tetrahedron Lett., 1996, 37, 1153–1156 CrossRef CAS; (b) M. Medow, G. Priem, G. Quelever, M. Camplo and J. K. Kraus, Tetrahedron Lett., 1998, 39, 4021 CrossRef; (c) Q. Zhao, L. Ma, S. Jiang, H. Lu, S. Liu, Y. He, N. Strick, N. Neamati and A. K. Debnath, Virology, 2005, 339, 213 CrossRef CAS PubMed; (d) F. Curreli, S. Choudhury, I. Pyatkin, V. P. Zagorodnikov, A. K. Bulay, A. Altieri, Y. D. Kwon, P. D. Kwong and A. K. Depnath, J. Med. Chem., 2012, 55, 4764–4775 CrossRef CAS PubMed.
- U. D. Treuner and H. Breuer, US Pat., 4, 1978, 113, 943, 1978transChem. Abstr., 1979, 90, 72212 Search PubMed.
- J. J. Hale, S. G. Mills, M. MacCoss, P. E. Finke, M. A. Cascieri, S. Sadowski, E. Ber, G. G. Chicchi, M. Kurtz, J. Metzger, G. Eiermann, N. N. Tsou, F. D. Tattersall, N. J. M. Rupniak, A. R. Williams, W. Rycroft, R. Hargreaves and D. E. Maclntyre, J. Med. Chem., 1998, 41, 4607 CrossRef CAS PubMed.
- (a) A. Baeyer and V. Drewson, Ber. Dtsch. Chem. Ges., 1882, 15, 2856 CrossRef; (b) J. Kamlet, Ind. Eng. Chem., 1944, 16, 362 CAS; (c) Comprehensive organic name reactions and reagents, ed. Z. Wang, Wiley: A John Wiley and sons, inc., 2009, vol. 1, pp. 136–139 Search PubMed.
- (a) D. J. Knobloch, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 15340 CrossRef CAS PubMed; (b) A. F. Khattab and T. Kappe, J. Chem. Res., 2006, 9, 609 CrossRef; (c) J. Protasiewicz and G. D. Mendenhall, J. Org. Chem., 1985, 50, 3220 CrossRef CAS; (d) Z. Du, W. B. Li, X. H. Zhu, F. Xu and Q. Shen, J. Org. Chem., 2008, 73, 8966 CrossRef CAS PubMed; (e) R. Neidlein and W. Lehr, Heterocycles, 1981, 16, 1179 CrossRef CAS; (f) M. L. Testa, E. Zaballos and R. J. Zaragoza, Tetrahedron, 2012, 68, 9583 CrossRef CAS; (g) W. Ziemkowska, E. Jaskowska, E. Zygadlo-Monikowska and M. K. Cyranski, J. Organomet. Chem., 2011, 696, 2079 CrossRef CAS.
- (a) N. Hadei, E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, J. Org. Chem., 2005, 70, 8503 CrossRef CAS PubMed; (b) D. P. Allen, M. M. V. Wingerden and R. H. Grubbs, Org. Lett., 2009, 11, 1261 CrossRef CAS PubMed; (c) M. Kuriyama, R. Shimazawa and R. Shirai, Tetrahedron, 2007, 63, 9393 CrossRef CAS; (d) S. Miljanic, L. Frkanec, Z. Meic and M. Zinic, Eur. J. Org. Chem., 2006, 1323 CrossRef CAS; (e) A. W. Waltman and R. H. Grubbs, Organometallics, 2004, 23, 3105 CrossRef CAS; (f) Y. A. Ibrahim, N. A. Al-Awadi, T. F. Al-Azemi and E. John, RSC Adv., 2014, 4, 38869 RSC.
- (a) N. Kambe, T. Inoue, T. Takeda, S. Fujiwara and N. Sonoda, J. Am. Chem. Soc., 2006, 128, 12650 CrossRef CAS PubMed; (b) T. Mizuno, M. Matsumoto and I. N. T. Hirashima, Synth. Commun., 2006, 23, 2139 CrossRef; (c) R. Richter, F. A. Stuber and B. Tucker, J. Org. Chem., 1984, 49, 3675 CrossRef CAS.
- A. R. Katritzky, D. P. M. Pleynet and J. R. Levell, Synthesis, 1998, 153 CrossRef.
- L. E. Kaim, L. Gaultier, L. Grimaud and E. Vieu, Tetrahedron Lett., 2004, 45, 8047 CrossRef.
- Z. Zhan, X. Cheng, X. Ma, J. Li, L. Hai and Y. Wu, Tetrahedron, 2015, 71, 6928 CrossRef CAS.
- (a) K. Stott, J. Stonehouse, J. Keeler, T.-L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1995, 117, 4199 CrossRef CAS; (b) K. Stott, J. Keeler, Q. N. Van and A. J. Shaka, J. Magn. Reson., 1997, 125, 302 CrossRef CAS.
- T. Bally and P. R. Rable, J. Org. Chem., 2011, 76, 4818 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr, R. E. Stratmann, J. C. Burant, et al., Gaussian 03, Revision A.6, Gaussian, Inc, Pittsburgh, PA, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR spectra and MALDI mass-spectra of the products 3a–h and 4a–h; crystallographic data for 4a,b (CIF), description of quantum chemical computational setup and comparison of X-ray and DFT computed structural parameters of 4a. CCDC 1417946 (for 4a) and 1015263 (for 4b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra02586b |
| This journal is © The Royal Society of Chemistry 2016 |