
Antimicrobial electrospun poly(ε-caprolactone) scaffolds for gingival fibroblast growth
Anna Baranowska-Korczyc*a,
Alicja Warowickaa,
Małgorzata Jasiurkowska-Delaportea,
Bartosz Grześkowiaka,
Marcin Jareka,
Barbara M. Maciejewskaab,
Justyna Jurga-Stopac and
Stefan Jurgaab
aNanoBioMedical Centre, Adam Mickiewicz University, Umultowska 85, PL-61614 Poznań, Poland. E-mail: akorczyc@amu.edu.pl
bDepartment of Macromolecular Physics, Faculty of Physics, Adam Mickiewicz University, Umultowska 85, PL-61614 Poznań, Poland
cDepartment of Biomaterials and Experimental Dentistry, Poznań University of Medical Sciences, Fredry 10, PL-61701 Poznań, Poland
First published on 10th February 2016
Abstract
This study discusses the value of polymer electrospun materials in three-dimensional (3D) scaffolds and antibacterial wound dressings for potential dental applications. Polycaprolactone (PCL) and polyvinylpyrrolidone (PVP) nanofibers were used as bases for gingival fibroblast (HGF-1 cell line) growth. HGF-1 cells cultured on both types of nanofibers were found to have normal morphology and growth by selective staining of the nuclei and cytoskeleton. The nanofibers were synthesized on different collectors to obtain a random or parallel alignment. Cell growth was observed along the nanofibers. In addition, antibiotics were incorporated within the nanofibers and studied by means of Raman spectroscopy and differential scanning calorimetry. The release profile of the antibiotics was determined by broad band dielectric measurements. The drug was found to be released by Fickian diffusion. The WST-1 test found PCL and PCL/ampicillin nanofibers to have minimal cytotoxicity. The antibacterial activity of materials containing ampicillin was evaluated by zone inhibition against a selected oral strain of Streptococcus sanguinis. The bacterial growth was inhibited by antibiotic release from PCL/ampicillin mats.
1. Introduction
In recent years, dental tissue engineering has focused on designing three-dimensional (3D), biocompatible, antimicrobial nanomaterials for wound dressing. Good wound healing membranes are characterized by high porosity and nanometer-size pores that protect them from bacterial infections and allow transport of gases and fluids to avoid bacterial invasion, pulp tissue inflammation, odontoblast death or dental trauma. Moreover, the field of regenerative endodontics needs a new 3D nanomaterial-based scaffold for dental tissue regeneration. A large range of nanostructures have been employed to give mechanical support for cell proliferation and differentiation, or have been combined into composite scaffolds such as polymer nanofibers (NFs),1–3 carbon nanotubes,4–6 zirconia nanoparticles,7 or titanium nanoalloys8,9 to improve their physio-chemical properties. However, their large surface to volume ratio, 3D structure and similarity to the fibrous architecture of the extracellular matrix (ECM) make electrospun nanofibrous scaffolds one of the most attractive materials for tissue engineering applications. The nanofibers can be molded into various shapes and forms such as porous structures, suitable for cell proliferation, differentiation and ingrowth.10 Additionally, the process provides the opportunity for direct encapsulation of drugs into the fibers to allow sustained release. Nanofibers containing an antibiotic can be used as a scaffold for regenerative endodontics without any symptoms of inflammation.11 The electrospun nanofibers can be synthesized using natural or synthetic polymers, including biodegradable ones. They are widely used in biological studies as in vitro scaffolds due to their high ability to integrate with surrounding native tissue.12,13Due to their hydrophilic nature and fibrous structure, polyvinylpyrrolidone (PVP) nanofibers are commonly used to form composite nanostructures as rapid delivery reagents for biological systems.14 Ferrocene/PVP nanofibers have been demonstrated to have antimicrobial activity against Gram-negative bacteria, with Escherichia coli as model organisms.15 Moreover, poly(dimethylsiloxane-b-PVP)-based fibrous scaffolds have been used as support for fibroblast adhesion, growth and proliferation.16
In contrast to hydrophilic nanofibers, water insoluble, biocompatible polymers are used to synthesize nanofibers which are stable in a biological environment, such as polycaprolactone (PCL). PCL is characterized by low toxicity, slow degradation time, water insolubility and low cost of synthesis.17 PCL nanofiber scaffolds and their composites are widely used for dental and orthopedic regenerative applications.18 They promote mineralization and tissue formation, and could be a good base for hard-tissue engineering applications. Fluorapatite incorporated PCL nanofibers provide a favorable extracellular matrix microenvironment for the growth, differentiation and mineralization of human dental pulp stem cells (DPSCs).19 PCL scaffolds whose hydrophilicity has been improved by electrospinning with PEO demonstrate enhanced cell infiltration, colonization and myofibroblastic differentiation.20 Electrospun PCL/gelatin membranes have also been assessed in in vivo studies as a preventive material for dura adhesions.21,22 Yang et al.23 report in vivo regeneration of hard tissue cultured on (PCL)/gelatin scaffolds.
In the present study, PCL and PVP electrospun nanofibers were prepared, characterized and then applied as scaffolds to facilitate gingival fibroblast (HGF-1 cell line) growth and to create a potential dental wound dressing with antibacterial activity. The interaction of the cells with nanostructures was studied depending on nanofiber stability and hydrophilicity. Selective staining of the nuclei and cytoskeleton revealed that the cultured HGF-1 cells demonstrated normal morphology and a growth oriented along the nanofibers for both types of scaffold. Antimicrobial activity was obtained by the incorporation of an antibiotic, ampicillin, into the nanofibers. The activity of the ampicillin-containing nanofibers against a selected oral strain of bacteria, Streptococcus sanguinis, was also studied. The ampicillin loading was analyzed by Differential Scanning Calorimetry (DSC) and its release by conductivity measurements. Moreover, the nanostructures were characterized by Scanning Electron Microscopy (SEM) and Raman spectroscopy. Our study indicates that nanofiber-based materials offer great potential as antimicrobial scaffolds for dental tissue engineering.
2. Materials and methods
2.1. Synthesis of PCL and PVP NFs
Poly(ε-caprolactone) (PCL) and polyvinylpyrrolidone (PVP) nanofibers were obtained by electrospinning. The starting solution of 10% wt PCL was composed of polycaprolactone (Mw = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and a mixture of chloroform and ethanol (3:1). The PVP (Mw = 360000) solution (12% wt) was prepared in ethanol. Both suspensions were left for a few days to obtain homogeneous solutions. During the electrospinning process, the flow rate was set as 1 ml h−1, and a 10 kV electric potential was maintained between the needle and the substrate. Three types of metallic collectors were used to obtain different alignment and amount of the nanofibers. A plate-shaped collector enabled the synthesis of randomly-oriented nanostructures, while a frame-shaped collector allowed the production of parallel-oriented NFs. The rotating collector (2500 rpm) allowed the production of nanofibrous mats. To obtain NFs with different concentrations of antibiotics, 5, 10 or 15 mg ampicillin sodium salt was added to 1 ml of both polymer solutions and sonicated for 0.5 h. All reactants were purchased from Sigma-Aldrich.
000) and a mixture of chloroform and ethanol (3:1). The PVP (Mw = 360000) solution (12% wt) was prepared in ethanol. Both suspensions were left for a few days to obtain homogeneous solutions. During the electrospinning process, the flow rate was set as 1 ml h−1, and a 10 kV electric potential was maintained between the needle and the substrate. Three types of metallic collectors were used to obtain different alignment and amount of the nanofibers. A plate-shaped collector enabled the synthesis of randomly-oriented nanostructures, while a frame-shaped collector allowed the production of parallel-oriented NFs. The rotating collector (2500 rpm) allowed the production of nanofibrous mats. To obtain NFs with different concentrations of antibiotics, 5, 10 or 15 mg ampicillin sodium salt was added to 1 ml of both polymer solutions and sonicated for 0.5 h. All reactants were purchased from Sigma-Aldrich.
2.2. Characterization of the NFs
The morphology of PCL and PVP nanofibers was investigated by SEM (Jeol 7001TTLS). Raman spectra of the electrospun nanofibers were obtained using a Renishaw spectrometer with an excitation laser wavelength of 785 nm. The electrospun nanofibers for SEM and Raman spectroscopy analysis were deposited on silicon substrates or as a nanofibrous mat.The nature of ampicillin in the NFs was assessed by specific heat measurements carried out by means of DSC on a DSC8000 (PerkinElmer) calorimeter. The calibration procedure was a multi-step process which involved optimizing the baseline, and then calibrating for sample temperature, furnace and heat flow. Reference indium and reference lead were used for temperature and enthalpy calibration. The DSC runs were recorded while heating and cooling the samples at a temperature rate of 10 K min−1. DSC spectra were collected for PCL, ampicillin, PCL NFs and PCL/ampicillin nanofibrous mats.
To estimate the quantity of drug released from the nanofibers in an aqueous environment, 24 hours after the synthesis process, the mat was placed in 25 ml of water, then the samples of soaking solution were collected for detection at pre-determined time points. The conductivity of solution was measured by means of Broad Band Dielectric Spectroscopy (BDS). The measurements were performed in the frequency range 1 Hz to 107 Hz at room temperature using a Novocontrol high-resolution alpha analyzer. The sample was mounted between parallel 10 mm diameter brass plates separated by a Teflon ring of a thickness of 8 mm. The complex dielectric function ε* = ε′(ω) + ε′′(ω), obtained by the BDS measurements, is equivalent to the complex conductivity function σ*(ω) = σ′(ω) − iσ′′(ω) where ω = 2πν = 2πT−1 is the angular frequency, with T as the time for one period. The real and imaginary part of σ*(ω) is given by σ′ = ε0ωε′′, σ′′ = ε0ωε′ where ε0 is dielectric permittivity of vacuum. The conductivity of the aqueous solution of the ampicillin sodium salt in the concentration range 0 to 0.5 mg ml−1 was measured to determine the concentration of the drug in the soaking solution. The percentage of the released drug was then determined based on the initial weight of the drug incorporated in the PCL nanofibers.
2.3. Gingival fibroblasts culture on PCL and PVP NFs
The human gingival fibroblast (HGF-1, American Type Culture Collection – ATCC, USA) cell line was cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS, Sigma-Aldrich), 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 1 mM sodium pyruvate. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. When the cell culture reached 80% confluence, cells were washed with phosphate buffer saline (PBS), trypsinized with trypsin–EDTA (Sigma-Aldrich) and counted using TC10™ automated cell counter (BioRad). To cultivate the HGF-1 cells on PCL and PVP nanofibers, the nanofibers were collected on glass slides, which were placed in MatTek plates (MatTek Corporation) and the cells were then seeded at a density of 1.5 × 105.2.4. Cell imaging by confocal microscopy
To study the growth and morphology of HGF-1 cells cultured on PCL and PVP nanofibers, the nuclei and cytoskeleton of the cells were stained. The cells were seeded in a MatTek plate at a density of 1.5 × 105 and incubated overnight at 37 °C. Prior to the staining procedure, the cells were fixed with 3.5% formaldehyde for 10 minutes and rinsed twice with PBS. In the next step, the cells were treated with 0.1% Triton X-100 for 5 minutes and also rinsed twice with PBS. The cells were subsequently treated with Oregon Green (Thermo Fisher Scientific's) dye at 20 μM concentration in 1% BSA solution for 20 minutes at room temperature for cytoskeleton staining. The cell nuclei were stained with red organic DRAQ5 (Cell Signaling) dye at a concentration of 5 μM for 5 minutes at room temperature. The cells were then rinsed twice with PBS. The samples were imaged using a confocal laser scanning microscope Olympus FV 1000. The 488 nm (for Oregon Green) and 635 nm (for DRAQ5) lasers were used for fluorescence excitation of HGF-1 cell organelles and the emitted light was detected in the range of 495–579 or 655–755 nm, respectively. The images were analyzed with FV10-ASW software (Olympus).2.5. Cell viability on antibiotic incorporated scaffolds
Cell viability on the PCL and PCL/ampicillin nanofibrous scaffolds was determined using a WST-1 Cell Proliferation Assay Kit (Clontech) with slight modification of the manufacturer's protocol. The HGF-1 cells were seeded on the mats at a density of 2.5 × 104 cells per well using a 12-well cell culture plate. After 72 hours, 25 μl of the WST-1 Cell Proliferation Reagent was added to each well and the cells were incubated for another 4 hours. Following this, 100 μl of supernatant was transferred to a well in a 96-well plate and the absorbance at 450 nm was recorded, using a multiwell plate reader (Zenyth, Biochrom). For this experiment, cell cultures growing in the well without any electrospun mat served as controls.2.6. Antimicrobial influence of the antibiotic incorporated NFs
The antimicrobial properties of the polymeric nanofibers incorporated with antibiotics were assessed against a model bacterial strain, Streptococcus sanguinis 2502 (Gram-positive, coccus-shaped), using a modified agar diffusion assay disc test. The S. sanguinis cells were spread on the solid surface of Brain Heart Infusion Agar (BHI Agar). Over these inoculated Petri dishes, small pieces of PCL nanofibers impregnated with ampicillin at the varying concentrations given above were distributed and incubated for 24 hours at 37 °C in an incubation chamber. After this period, the observed inhibition zones were photographed.3. Results and discussion
In this investigation, PCL and PVP nanofibers were synthesized by electrospinning. The electrospinning technique allowed single nanofibers and mats composed of entwined polymer quasi-one dimensional (1D) nanostructures to be obtained. Fig. 1 presents PCL and PVP nanofibers synthesized on two different types of collectors. The nanostructures were collected on a metallic plate to obtain randomly-orientated nanofibers and on a metallic frame base to obtain parallel-oriented nanofibers. The metallic plate collector provided an even distribution of electric carriers and attracted the polymer jet equally across the whole surface, resulting in a random alignment of the nanofibers (Fig. 1a and b). This alignment produced a highly porous material with nanometer-sized spaces. The frame shape-collector (10 × 10 mm – sample size, inset in Fig. 1c) allowed randomly-oriented nanofibers to be produced (Fig. 1c and d and inset in 1d). This is a commonly-used method in electronics and optoelectronics as nanostructures with a parallel orientation can be used in the construction of various devices.24 Li et al. report that the alignment and assembly of the nanofiber can be controlled by the shape and pattern of the collector, as a result of the tendency of nanofibers to be oriented along the direction along which minimizes the net torque of electrostatic forces applied to the two ends of the collected fiber.25 | ||
| Fig. 1 SEM images of randomly (a, b) and parallel (c, d) oriented PCL (a, c) and PVP (b) nanofibers. Inset (c) and (b) the frame-shaped collector for synthesizing parallel-aligned nanofibers. | ||
Moreover, the nanofibers were collected on the rotating collector to synthesize PCL and PVP nanofibrous mats (Fig. 2, inset). This is an efficient and effective way to produce electrospun non-woven materials. Fig. 2 shows PCL nanofibers collected for 20 minutes as a non-woven mat, whose size is defined by the length and diameter of the drum. The mat can be easily cut into smaller pieces, with a desired shape and size. The nanofibrous mat can be used in wound dressings or bandages for medical purposes.
 | ||
| Fig. 2 A mat composed of PCL nanofibers. Inset: The setup with a rotating collector for synthesis of nanofibrous mats. | ||
To obtain antimicrobial electrospun materials for potential dental wound dressings, various concentrations of ampicillin were incorporated into the mats by addition to the initial solutions. The mats were produced using a rotating collector to obtain tubular structures, which were then further cut and removed from the drum. Fig. 3 presents the outer side of the PCL and PVP mats with antibiotics. The mats formed dense fibrous structures due to the overlapping of the subsequent layers. This is consistent with previous reports, which describe electrospun tubular structures as multilayered conduits with aligned fibers in the inner layer and non-woven random mats in the outer layer.26 This method of synthesis is commonly used for the preparation of tubular structures in vascular graft engineering due to their resemblance to the structure of natural vessels, their good mechanical properties, and the way the provide a suitable environment for cell growth, proliferation and differentiation.27,28
 | ||
| Fig. 3 SEM images of PVP (a, b) and PCL (c, d) nanofibrous mats, incorporated 5 mg ml−1 (a, c) and 15 mg ml−1 (b, d) ampicillin and synthetized on drum collector. The images show outer layer of the mats. | ||
Raman spectroscopy is a versatile tool for studying two aspects of molecular systems: (i) the presence of conformational isomerism from a band position assignment, and (ii) the degree of crystalline and amorphous phase from assigned peaks. Fig. 4a presents the Raman spectra of the electrospun PCL fibers and ampicillin incorporated PCL nanofibers. Several narrow peaks at 913 cm−1 (νC–COO), and others within the spectral ranges 1003–1110 cm−1 (skeletal stretching), 1270–1320 cm−1 (ωCH2), 1405–1470 cm−1 (δCH2) and 2800–3200 cm−1 (νCH) are referred to the crystalline fraction.29–32 The broad peak at 865 cm−1 indicates that the amorphous phase is also present in PCL nanofibers. In order to determine the degree of the crystallinity of the sample, a band in the region 1710–1750 cm−1 (corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) was deconvoluted into two Gaussian lines centered at 1724 cm−1 and 1732 cm−1 (see Fig. 4b). The first peak corresponds to crystalline phase whereas the latter is ascribed to amorphous domains. The fraction of crystalline phase Xc was calculated according to the equation: Xc = Ic/(Ic + Ia) where Ic and Ia refer to the integrated intensity of crystalline and amorphous components.30 Xc was found to be around 41%.
O stretching) was deconvoluted into two Gaussian lines centered at 1724 cm−1 and 1732 cm−1 (see Fig. 4b). The first peak corresponds to crystalline phase whereas the latter is ascribed to amorphous domains. The fraction of crystalline phase Xc was calculated according to the equation: Xc = Ic/(Ic + Ia) where Ic and Ia refer to the integrated intensity of crystalline and amorphous components.30 Xc was found to be around 41%.
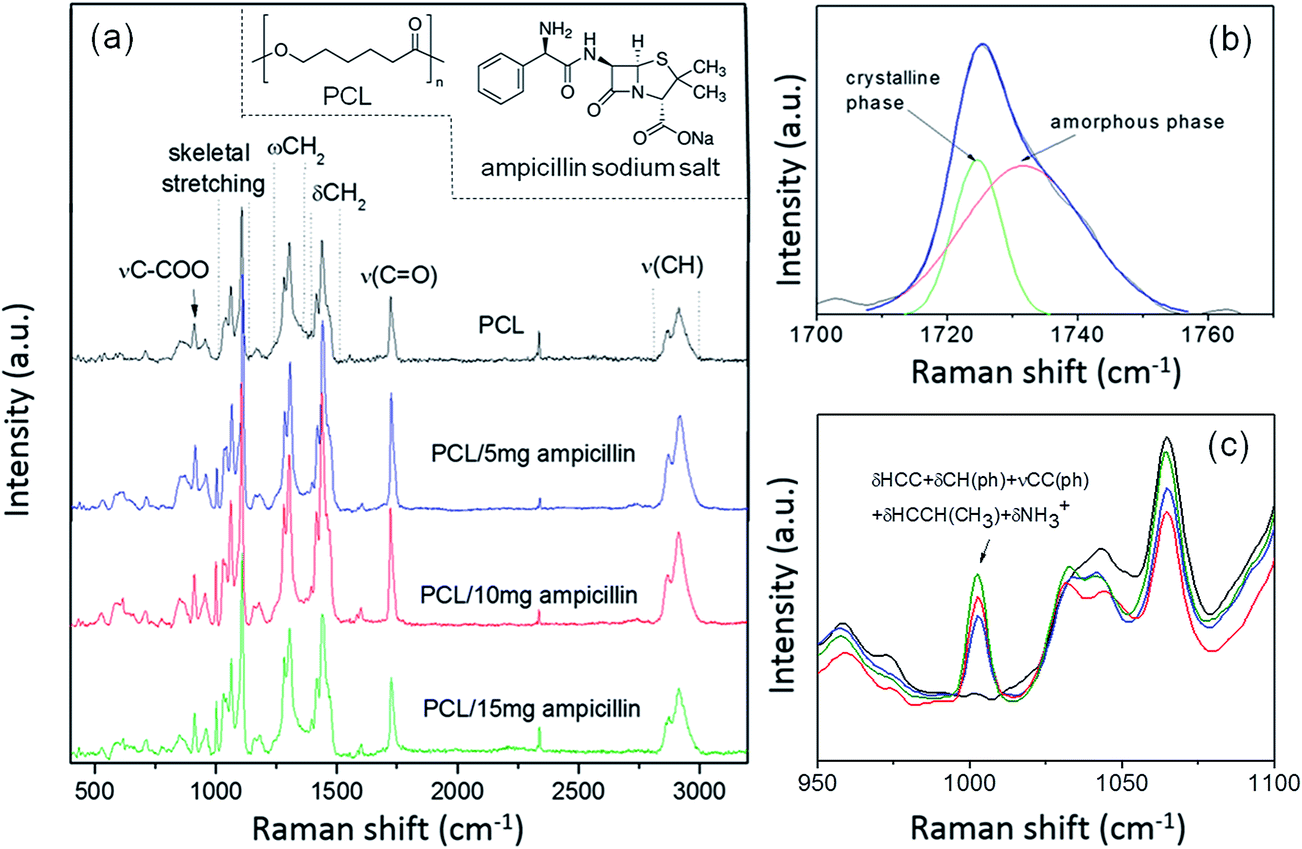 | ||
| Fig. 4 Raman spectra of electrospun PCL and PCL/ampicillin nanofibers (a), CO stretching (νCO) range showing the contribution of amorphous and crystalline fractions (b), the spectral region displaying the combination mode related to ampicillin (c). Inset (a) the chemical formula of PCL and ampicillin sodium salt. | ||
The crystallinity of the matrix plays an important role on drug release profiles, with a crystalline matrix releasing the drug at a much slower rate than its amorphous counterpart of similar molecular weight.33 Jeong et al.34 report the effects of the presence of a crystalline microstructure on the drug release behavior of poly(ε-caprolactone) microspheres. They note that the release of papaverine from PCL microparticles is controlled by drug diffusion through the amorphous region of the polymer matrix, not by polymer erosion. Moreover, PCL microstructures with a more crystalline structure exhibit a tendency to more sustained drug release. In our study, the amorphous phase of PCL was estimated to be about 60%, which indicates the release profile of ampicillin is fast, especially in the first stage of the process. The Raman spectra also indicate the presence of ampicillin in the PCL nanofibers, with the presence of a band corresponding to the combination mode in ampicillin at 1003 cm−1 (Fig. 4c).
DSC studies were performed to analyze the thermal transitions of PCL and composite nanofibers. Fig. 5 shows the thermograms for PCL, ampicillin and the PCL composite nanofibers as a function of ampicillin concentrations. An endothermic melting peak around 56 °C, characteristic for polycaprolactone,35 was found to be present for the polymer as a substrate of the synthesis (Fig. 5a), as well as PCL/ampicillin (Fig. 5c–e) and pure PCL (Fig. 5f) nanofibers. The thermogram for ampicillin reveals a low intensity and a broad endothermic peak centered around 68 °C corresponding to water loss and also indicating weakly-bound water molecules (Fig. 5b).36
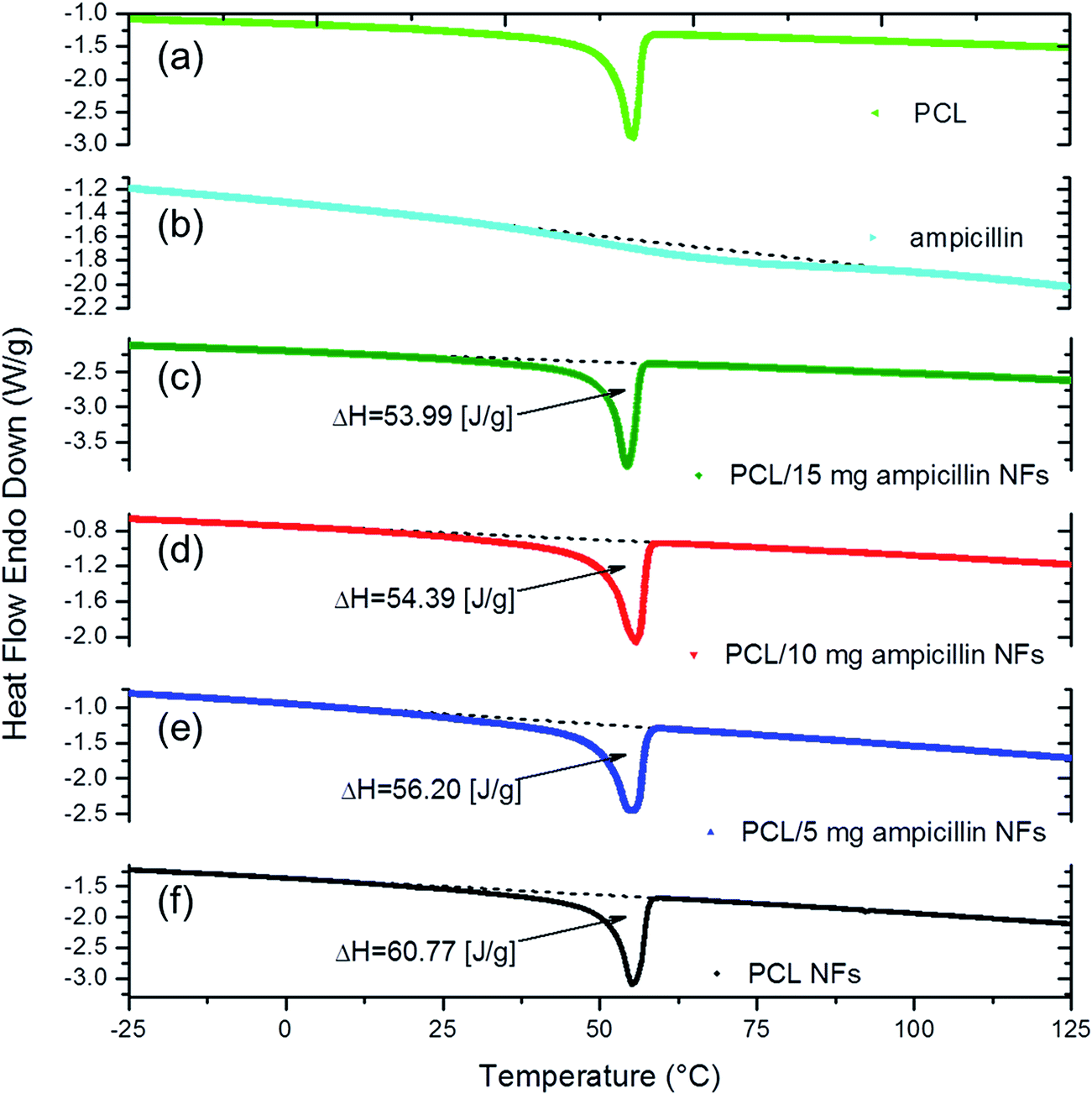 | ||
| Fig. 5 DSC thermograms of PCL (a), ampicillin (b) and PCL composite nanofibers loaded with 15 (c), 10 (d), 5 (e) and 0 (f) mg of ampicillin. | ||
To elucidate the degree of NF crystallinity and content of the loaded antibiotic within the nanostructures, the enthalpy value (ΔH) for the PCL peak around 56 °C was determined for all synthetized composite PCL/ampicillin and pure PCL nanofibers. The values were estimated to be 53.99, 54.39, 56.20 and 60.77 J g−1 for PCL/15 mg ampicillin, PCL/10 mg ampicillin, PCL/5 mg ampicillin and pure PCL nanofibers, respectively (Fig. 5c–f). The melting enthalpy of PCL NFs increased as a result of decreasing the concentration of ampicillin in the nanostructures.35 The highest ΔH value was noted for pure polymer nanofibers, which indicated that increasing the antibiotic concentration inside the NFs caused a reduction of crystalline phase content in the NFs. It is important to note that the experimental conditions were carefully controlled to avoid any loss of material, such as by the production of the NFs outside the collector.
It can be concluded that all the starting solution was used for NF production, with no loss, and the weight ratio of the substrates, polymer to antibiotic, did not change significantly after the synthesis process. DSC analysis was used as a qualitative verification of the electrospinning process, which provides quasi 1D nanostructures made of a composition of substrates without a solvent, which evaporated during the process, resulting in the solidification of all loaded components.37 Several reports also indicate that the electrospinning process does not affect the activity of different compounds incorporated into the NFs, such as drugs,38 proteins39 or even cells.40
The amount of released drug in this studies can not be determined by commonly used the UV-Vis spectrometric method because ampicillin does not show any absorption peak in the visible and ultraviolet wavelength range. Therefore we used the fact that released sodium salt ampicillin changes the conductivity of soaking solution. The release of the antibiotic from the PCL nanofibers in water was monitored by conductivity measurements as described in Experimental section. The real part of the complex conductivity versus frequency σ′(ν) obtained at selected time points for the aqueous solution with the soaked mat as well as the aqueous solution of sodium salt ampicillin is presented in Fig. 6. The frequency dependence of σ′(ν) of the studied solutions is characterized on the intermediate frequency regime by a plateau, the value of which corresponds with the dc conductivity. At low frequencies, σ′(ν) shows decreasing trend due to electrode polarization. As the ampicillin sodium salt is dissolved in water, the number of positive and negative ions increases in the solution. Consequently, the dc conductivity of the aqueous solution also increases. The dc conductivity of the aqueous solution with the soaked PCL/15 mg ampicillin mat is shown in Fig. 7a as a function of soaking time. Based on the calibration curve given in Fig. 7b and the initial quantity of antibiotic incorporated into polymer nanofibers, the percentage of released ampicillin can be calculated (see Fig. 7c). The time for half the total amount of the drug to be released from the fibres is 3.4 hours. To find mechanism of drug release, the data given in Fig. 7c were analysed using the Korsmeyer–Peppas model, Mt = Ktn, where Mt is a fraction of the drug released at time t, K is a release rate constant and n is a parameter which characterizes the release mechanism. The value n = 2.3 obtained for the percentage of ampicillin released from the PCL nanofibers indicates a Fickian diffusion mechanism.41
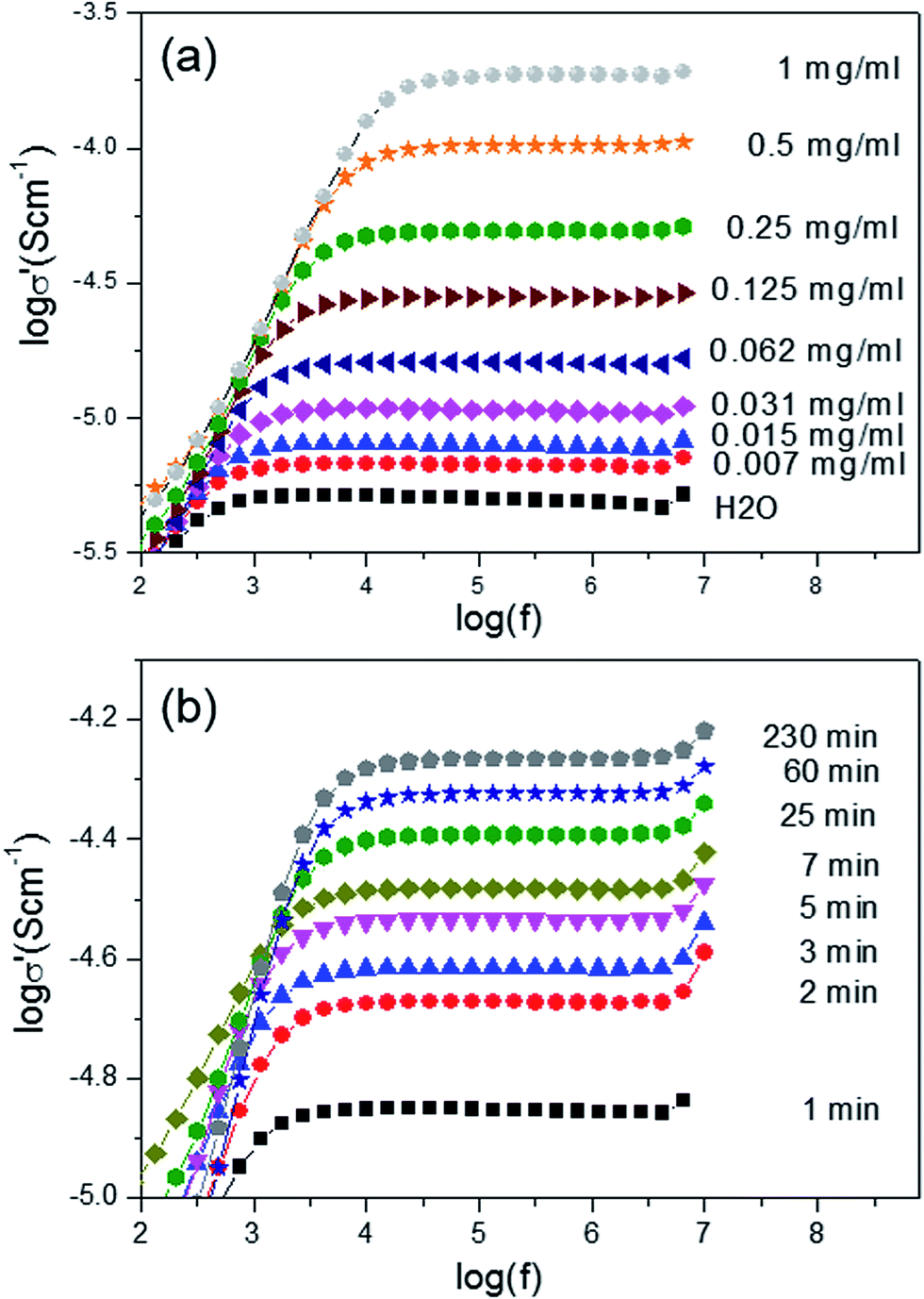 | ||
| Fig. 6 The real part of the complex conductivity versus frequency σ′(ν) obtained at selected time points for the aqueous solutions with the soaked mat (a) and the aqueous solution of the ampicillin sodium salt (b) in the concentration range 0 to 1 mg ml−1. | ||
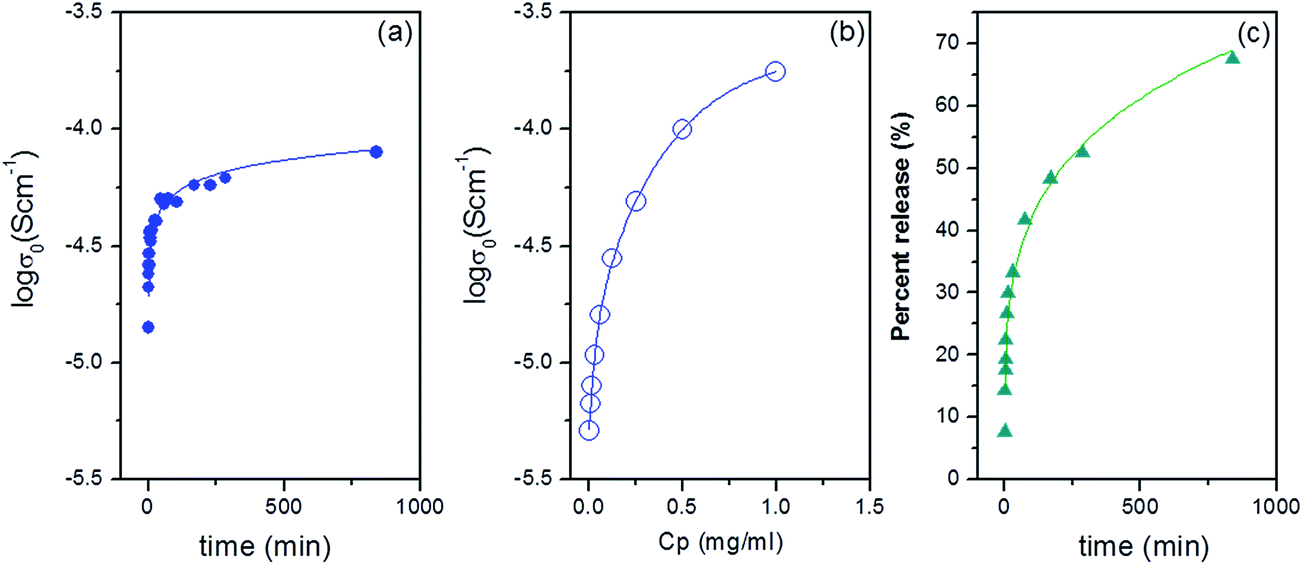 | ||
| Fig. 7 The dc conductivity σ0 of the aqueous solution with the soaked PCL/15 mg ampicillin mat (a) dc conductivity σ0 of the water–ampicillin solution in the concentration range 0 to 1 mg ml−1 (calibration curve) (b), ampicillin release profile from PCL nanofibers (c). | ||
Fig. 8a presents confocal images of HGF-1 cells cultured on PCL nanofibers. The cells were stained with a green organic dye for visualizing the cytoskeleton and red dye for the nuclei. HGF-1 cells demonstrated a normal morphology and growth affected by fiber alignment. The nanofibers were collected randomly for these experiments. The cells grew between the nanofibers and attached to them as a base. The morphology of the nuclei did not differ from that of control samples (Fig. 8b). The cytoskeleton of the cells was spread between the fibers and seems to be less developed in comparison to control samples (Fig. 8b), although the light and confocal microscope analyses indicate the morphology of the cells and cytoskeleton to be normal. We conclude that cytoskeleton network depends on the nanofiber number, and their system depends on the substrate. The cells in the control sample grew only on the substrate and their cytoskeleton network spread in all directions. In case of the 1D nanostructure support, the growth of the cells is controlled by their alignment. Moreover, nanofibers form a 3D scaffold and mimic the natural environment of the cells. Owing to its good biodegradability and mechanical properties, which are suitable for tissue engineering, PCL is one of the most commonly-applied electrospun materials in biological sciences.42
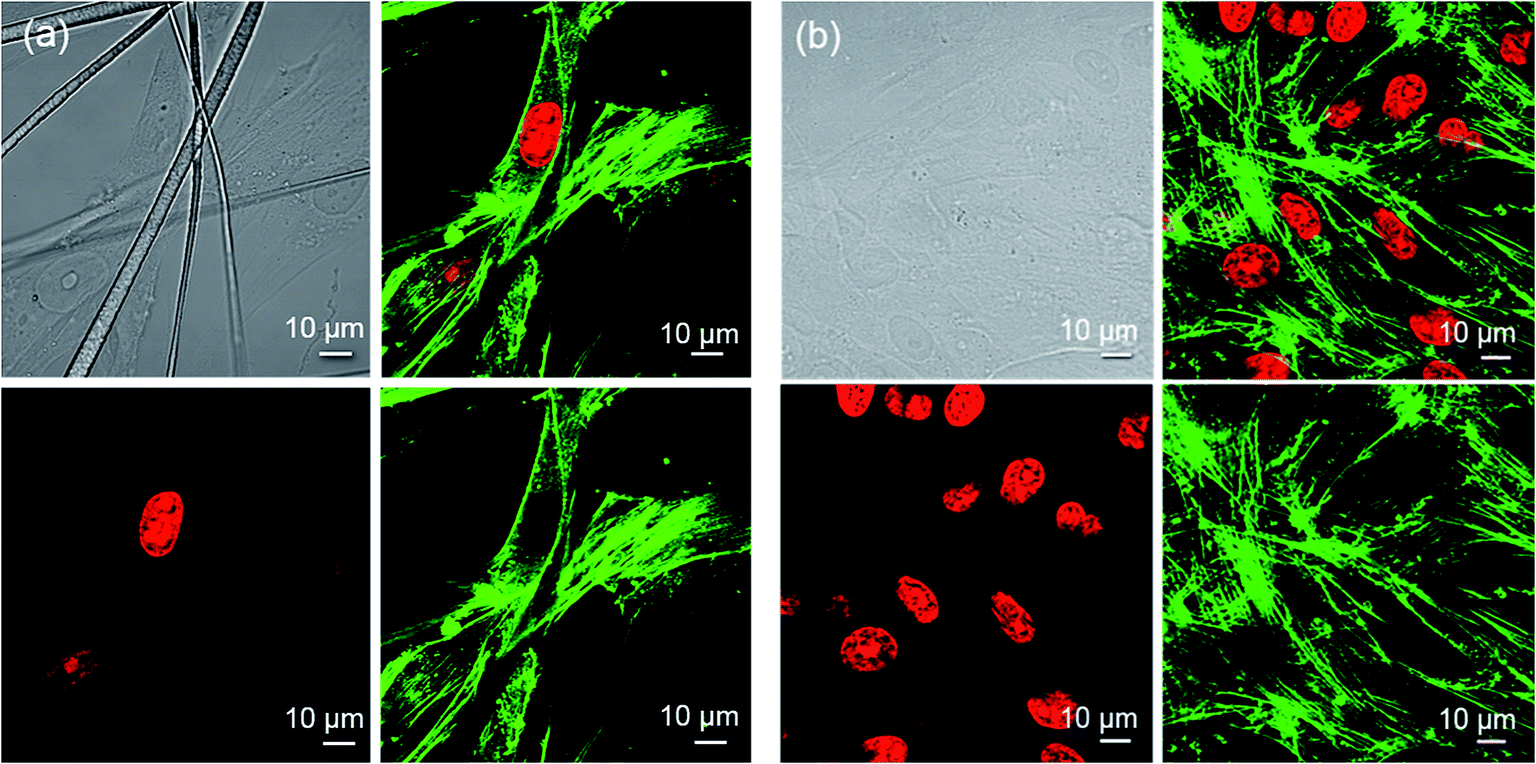 | ||
| Fig. 8 Confocal images of HGF-1 cells grown on PCL nanofibers (a) and MatTek plate – control sample (b) after cytoskeleton (green) and nuclei (red) staining. | ||
PCL was used in the present study due to its high biocompatibility, high biodegradability, and high toughness. The relatively slow biodegradability of PCL enables the drug release to be studied in detail. However, its hydrophobicity causes poor surface wetting, as well as poor cell adhesion and proliferation, and therefore is typically combined with other more hydrophilic polymers.43 Our results indicate that PCL nanofibers have high resistance and biocompatibility, as well as an influence on the growth of human gingival fibroblasts. These findings are consistent with several other reports examining the effect of PCL nanofibers on the growth and morphology of several other cell types.26,44 Cell proliferation tests found PCL nanofibers to be biocompatible with human dermal fibroblasts, even more so when the fibers were coated by collagen.45 PCL/poly(dopamine) composed nanofibers have also been used as a scaffold to mimic the adhesive properties of mussels, thus promoting the adhesion and viability of human umbilical vein endothelial cells.46 However, mixing PCL with other substances can lead to a reduction in the mechanical strength of the nanofiber,43 and make them unstable within a biological environment for the long term. Another biocompatible hydrophilic polymer, PVP, was chosen to compare its cell interaction with that of the hydrophobic PCL.
The HGF-1 cells cultured on PVP nanofibers also revealed normal morphology and growth along the nanostructures (Fig. 9a), with normal shape and number of the nuclei. However, the cytoskeleton network is less developed than for PCL nanofibers. We conclude that this is a result of the low stability of the nanofibers in the biological environment. Fig. 9 presents a stained PVP NF sample, with Fig. 9b showing the cells at a higher density than in Fig. 9a, this being needed to obtain a sufficient quantity of cells for detailed morphological study. Fig. 9b shows a bright light image of the cells growing in a defined direction, in contrast to the previously synthesized nanostructures, despite the nanofibers themselves being completely dissolved. This suggests that PVP nanofibers can control the cell alignment in the first phase of growth, but they are not sufficiently resistant to in vitro culture conditions. Preferential growth along fibers has also previously been observed for neurites, although in this case, the PCL nanofibers were covered with a laminin layer to allow more effective guidance.47 Our findings indicate that pure PVP and PCL nanofibers can significantly control HGF-1 cell growth. However, PCL nanofibers are more suitable than PVP nanostructures for forming scaffolds for biological applications due to their high stability in liquids. For this reason, PCL nanofibers were selected to form wound dressing mats with integrated antibiotics. Ampicillin/PVP nanofibrous mats dissolved very quickly in the tested medium and they are not suitable for designing sustained release nanomaterials.
 | ||
| Fig. 9 Confocal images of HGF-1 grown on PVP nanofibers after cytoskeleton (green) and nuclei (red) staining: high (a) and low (b – with cell alignment) magnification. The images present low (a) and high (b) density PVP NF samples. | ||
A WST-1 test was performed to provide a quantitative verification of the influence of pure PCL NF and PCL nanofibers with incorporated ampicillin on HGF-1 cell viability. Fig. 10 indicates that of PCL NFs as well as 5, 10 and 15 mg ml−1 ampicillin loaded nanostructures demonstrated a minimal cytotoxic effect in comparison to the viability of the cells growing without nanofibers (control sample). The level of cell viability for the pure NF culture is comparable with that of the antibiotic-containing samples. Neither the pure PCL NFs nor the nanofibers with antibiotics were found to affect cell growth. No substantial down-regulation in HGF-1 cell viability was found in any antibiotic-treated NFs across the whole applied range of ampicillin concentrations, which indicates that the antimicrobial scaffold for gingival fibroblast growth can be designed based on electrospun nanomaterials. Moreover, PCL/ampicillin NF scaffolds can prevent microorganism growth due to both the presence of antibiotics and the presence of small nanosize pores.
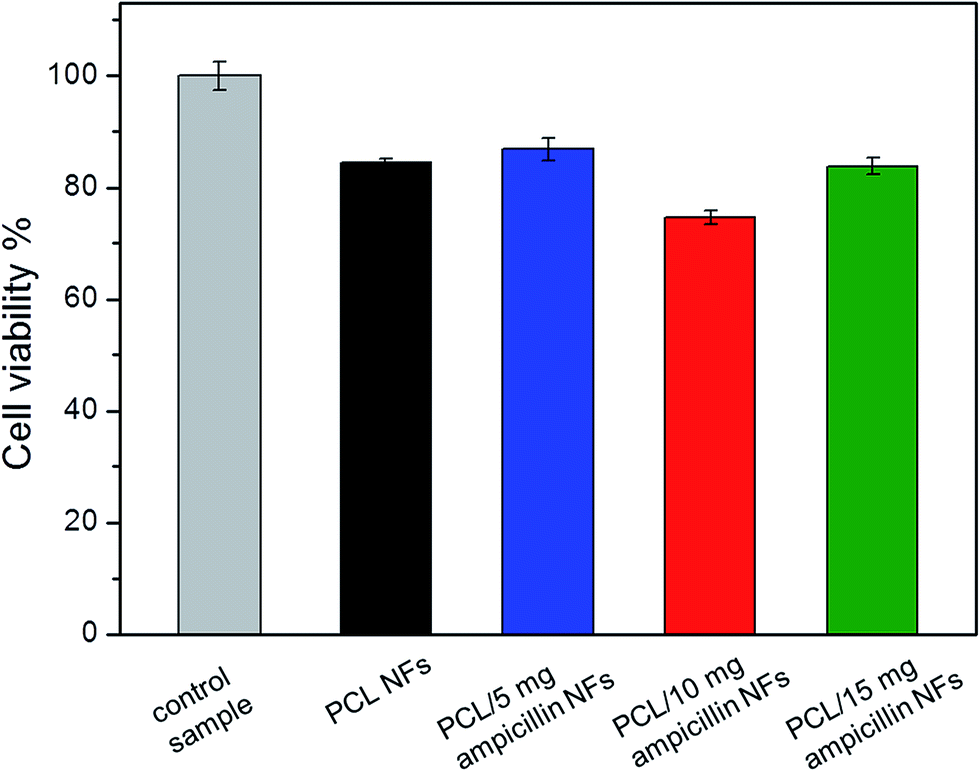 | ||
| Fig. 10 HGF-1 cell viability in terms of respiration activity measured 72 hours after seeding the cells on PCL nanofibers incorporated 0 mg ml−1 (pure PCL NFs), 5 mg ml−1, 10 mg ml−1 and 15 mg ml−1 ampicillin. Cells growing without PCL NFs were used as a control sample. | ||
The antimicrobial activity of PCL/ampicillin and PVP/ampicillin nanofibers was examined in solid culture medium. PCL and PVP nanofibrous mats incorporating antibiotic were placed on Streptococcus sanguinis culture. The nanofibers and pure ampicillin (control sample) were incubated for 24 hours. PCL nanofibers were found to be stable during the experiments, in contrast to the PVP NFs, which dissolved very quickly. Our findings indicate that compared to the PVP nanostructures, PCL NFs are more suitable for forming scaffolds for biological application due to their high stability in a biological environment. As they are hydrophilic, PVP NFs are not suitable for the design of long-term and sustained-release nanomaterials in biological liquids. Fig. 11 shows the zones of inhibition produced by the ampicillin and all synthetized PCL/ampicillin mats. The growth inhibition zone was increased as the antibiotic loading inside the materials increased.
 | ||
| Fig. 11 Activity of PCL/ampicillin nanofibers against S. sanguinis depicting zones of inhibition of ampicillin – control (1) pure PCL mat (2) and PCL/ampicillin mats loaded with 5 mg ml−1 (3), 10 mg ml−1 (4) and 15 mg ml−1 (5) of antibiotic. | ||
Despite the fact that it is usually found in the dental plaque of the human oral cavity, S. sanguinis was selected for our studies as its presence in the blood stream, e.g. after dental surgery, can cause acute bacterial endocarditis and further colonization of the heart valves.48 To avoid infection and systemic treatment, nanofibrous biocompatible polymer/antibiotic mats can be applied as wound dressings in dental treatment. As dental wound dressings, antibiotic-containing electrospun mats can be used for local treatment, as they affect S. sanguinis close to the site of the injury. Moreover, mats with a nanoporous structure provide a natural barrier to protect against microorganisms. The PCL/ampicillin nanofibers can also be used to form antimicrobial 3D scaffolds for gingival fibroblast growth due to their low toxic effect. The biodegradable and biocompatible scaffolds mimic the natural environment of HGF-1 cells and allow new dental materials, drugs and treatments to be tested.
4. Conclusions
The PCL and PVP electrospun nanofibers presented in this report were applied as human gingival fibroblast (HGF-1) scaffold and antimicrobial membranes for potential dental purposes. The nuclei and cytoskeleton morphology of HGF-1 cells grown on both types of nanofibers was unchanged. Hydrophilic PVP nanofibers demonstrated low stability in a biological environment in comparison to hydrophobic PCL nanofibers. Moreover, the direction of cell growth was governed by nanofiber number and alignment. PCL/ampicillin mats were successfully prepared using a drum collector. The composed nanofibers were studied by Raman spectroscopy and DSC analysis. Conductivity measurements of the soaking solution using Broad Band Dielectric methods found the mechanism of drug release to be based on Fickian diffusion, and the WST-1 test found the PCL and PCL/ampicillin nanofibers to have minimal cytotoxicity. The mats released antibiotics and showed antibacterial activity against a selected dental caries pathogen. The zone of growth inhibition for Streptococcus sanguinis was directly proportional into the content of ampicillin incorporated to the mats. Our findings indicate that the electrospun nanofibers are promising materials for dental engineering and wound dressing with the ability to reduce oral pathogens.Acknowledgements
The research was supported by the National Science Centre (Grant SONATA6: UMO-2013/11/D/ST5/02900), the European Social Fund (POKL.04.03.00-00-015/12) and Nation Centre for Research and Development (Grant: PBS1/A9/13/2012).References
- M. T. P. Albuquerque, M. C. Valera, M. Nakashima, J. E. Nor and M. C. Bottino, J. Dent. Res., 2014, 93, 1222–1231 CrossRef CAS PubMed.
- G.-H. Kim, Y.-D. Park, S.-Y. Lee, A. El-Fiqi, J.-J. Kim, E.-J. Lee, H.-W. Kim and E.-C. Kim, J. Biomater. Appl., 2014, 29, 854–866 CrossRef PubMed.
- X. He, L. Cheng, X. Zhang, Q. Xiao, W. Zhang and C. Lu, Carbohydr. Polym., 2015, 115, 485–493 CrossRef CAS PubMed.
- F. M. P. Tonelli, A. K. Santos, K. N. Gomes, E. Lorençon, S. Guatimosim, L. O. Ladeira and R. R. Resende, Int. J. Nanomed., 2012, 7, 4511–4529 CAS.
- A. Abarrategi, M. C. Gutiérrez, C. Moreno-Vicente, M. J. Hortigüela, V. Ramos, J. L. López-Lacomba, M. L. Ferrer and F. del Monte, Biomaterials, 2008, 29, 94–102 CrossRef CAS PubMed.
- H. Wang, C. Chu, R. Cai, S. Jiang, L. Zhai, J. Lu, X. Li and S. Jiang, RSC Adv., 2015, 5, 53550–53558 RSC.
- L. Bao, J. Liu, F. Shi, Y. Jiang and G. Liu, Appl. Surf. Sci., 2014, 290, 48–52 CrossRef CAS.
- P. Jiang, J. Liang and C. Lin, Appl. Surf. Sci., 2013, 280, 373–380 CrossRef CAS.
- M. Sowa, M. Piotrowska, M. Widziołek, G. Dercz, G. Tylko, T. Gorewoda, A. M. Osyczka and W. Simka, Mater. Sci. Eng., C, 2015, 49, 159–173 CrossRef CAS PubMed.
- S. Agarwal, J. H. Wendorff and A. Greiner, Polymer, 2008, 49, 5603–5621 CrossRef CAS.
- M. C. Bottino, K. Kamocki, G. H. Yassen, J. A. Platt, M. M. Vail, Y. Ehrlich, K. J. Spolnik and R. L. Gregory, J. Dent. Res., 2013, 92, 963–969 CrossRef CAS PubMed.
- F. K. K. Wan-Ju Li, C. T. Laurencin, E. J. Caterson and R. S. Tuan, J. Biomed. Mater. Res., 2002, 60, 613–621 CrossRef PubMed.
- K. Gupta, A. Haider, Y. Choi and I. Kang, Biomater. Res., 2014, 18, 5 CrossRef PubMed.
- M. Dai, S. Jin and S. R. Nugen, Biosensors, 2012, 2, 388–395 CrossRef CAS PubMed.
- J. H. Chai and Q. S. Wu, Beilstein J. Nanotechnol., 2013, 4, 189–197 CrossRef CAS PubMed.
- I. L. Keranov, M. Michel, A. Kostadinova, V. Toniazzo, D. Ruch and T. Vladkova, Open J. Biophys, 2013, 2013, 148–157 CrossRef.
- D. J. M. L. D. Shea, D. Wang and R. T. Franceschi, Tissue Eng., 2000, 6, 605–617 CrossRef PubMed.
- L. Zhang, Y. Morsi, Y. Wang, Y. Li and S. Ramakrishna, Japanese Dental Science Review, 2013, 49, 14–26 CrossRef.
- T. Guo, Y. Li, G. Cao, Z. Zhang, S. Chang, A. Czajka-Jakubowska, J. E. Nor, B. H. Clarkson and J. Liu, J. Dent. Res., 2014, 93, 1290–1295 CrossRef CAS PubMed.
- Y. F. Li, H. Gregersen, J. V. Nygaard, W. Cheng, Y. Yu, Y. Huang, M. Dong, F. Besenbacher and M. Chen, Nanoscale, 2015, 7, 14989–14995 RSC.
- R. Shi, J. Xue, H. Wang, R. Wang, M. Gong, D. Chen, L. Zhang and W. Tian, J. Mater. Chem. B, 2015, 3, 4063–4073 RSC.
- H. Yoshimoto, Y. M. Shin, H. Terai and J. P. Vacanti, Biomaterials, 2003, 24, 2077–2082 CrossRef CAS PubMed.
- X. Yang, F. Yang, X. F. Walboomers, Z. Bian, M. Fan and J. A. Jansen, J. Biomed. Mater. Res., Part A, 2010, 93, 247–257 Search PubMed.
- A. Baranowska-Korczyc, K. Fronc, Ł. Kłopotowski, A. Reszka, K. Sobczak, W. Paszkowicz, K. Dybko, P. Dłuzewski, B. J. Kowalski and D. Elbaum, RSC Adv., 2013, 3, 5656–5662 RSC.
- D. Li, G. Ouyang, J. T. McCann and Y. Xia, Nano Lett., 2005, 5, 913–916 CrossRef CAS PubMed.
- X. Wang, B. Ding and B. Li, Mater. Today, 2013, 16, 229–241 CrossRef CAS PubMed.
- Y. Liu, K. Xiang, H. Chen, Y. Li and Q. Hu, AIP Adv., 2015, 5, 041318 CrossRef.
- W. Liu, S. Thomopoulos and Y. Xia, Adv. Healthcare Mater., 2012, 1, 10–25 CrossRef CAS PubMed.
- M. A. Ermeydan, E. Cabane, P. Hass, J. Koetz and I. Burgert, Green Chem., 2014, 16, 3313 RSC.
- O. Hartman, C. Zhang, E. L. Adams, M. C. Farach-Carson, J. Petrelli, B. D. Chase and J. F. Rabolt, Biomaterials, 2010, 31, 5700–5718 CrossRef CAS PubMed.
- G. Kister, G. Cassanas, M. Bergounhon, D. Hoarau and M. Vert, Polymer, 2000, 41, 925–932 CrossRef CAS.
- P. Taddei, A. Tinti and G. Fini, J. Raman Spectrosc., 2001, 32, 619–629 CrossRef CAS.
- A. Frank, S. K. Rath and S. S. Venkatraman, J. Controlled Release, 2005, 102, 333–344 CrossRef CAS PubMed.
- J.-C. Jeong, J. Lee and K. Cho, J. Controlled Release, 2003, 92, 249–258 CrossRef CAS PubMed.
- M. Zamani, M. Morshed, J. Varshosaz and M. Jannesari, Eur. J. Pharm. Biopharm., 2010, 75, 179–185 CrossRef CAS PubMed.
- C. Baraldi, A. Tinti, S. Ottani and M. C. Gamberini, J. Pharm. Biomed. Anal., 2014, 100, 329–340 CrossRef CAS PubMed.
- H. Li, Y. Xu, H. Xu and J. Chang, J. Mater. Chem. B, 2014, 2, 5492–5510 RSC.
- J. Ma, J. Meng, M. Simonet, N. Stingelin, A. A. J. M. Peijs and G. B. Sukhorukov, J. Mater. Sci.: Mater. Med., 2015, 26, 205 CrossRef CAS PubMed.
- T. Kowalczyk, A. Nowicka, D. Elbaum and T. A. Kowalewski, Biomacromolecules, 2008, 9, 2087–2090 CrossRef CAS PubMed.
- S. N. Jayasinghe, Analyst, 2013, 138, 2215–2223 RSC.
- S. Dash, P. N. Murthy, L. Nath and P. Chowdhury, Acta Pol. Pharm., 2010, 67, 217–223 CAS.
- G. H. Kim, Biomed. Mater., 2008, 3, 025010 CrossRef PubMed.
- L. Ghasemi-Mobarakeh, M. P. Prabhakaran, M. Morshed, M.-H. Nasr-Esfahani and S. Ramakrishna, Biomaterials, 2008, 29, 4532–4539 CrossRef CAS PubMed.
- M.-C. Chen, Y.-C. Sun and Y.-H. Chen, Acta Biomater., 2013, 9, 5562–5572 CrossRef CAS PubMed.
- Y. Z. Zhang, J. Venugopal, Z. M. Huang, C. T. Lim and S. Ramakrishna, Biomacromolecules, 2005, 6, 2583–2589 CrossRef CAS PubMed.
- S. H. Ku and C. B. Park, Biomaterials, 2010, 31, 9431–9437 CrossRef CAS PubMed.
- J. Xie, M. R. MacEwan, X. Li, S. E. Sakiyama-Elbert and Y. Xia, ACS Nano, 2009, 3, 1151–1159 CrossRef CAS PubMed.
- A. Dziedzic, R. Wojtyczka and R. Kubina, Molecules, 2015, 20, 13705–13724 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |