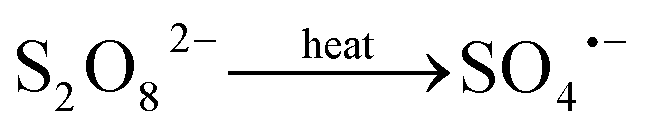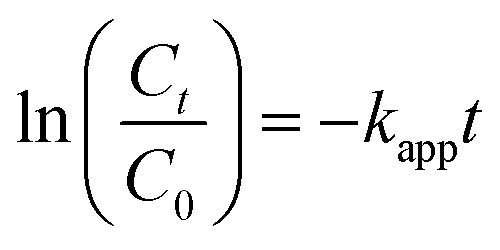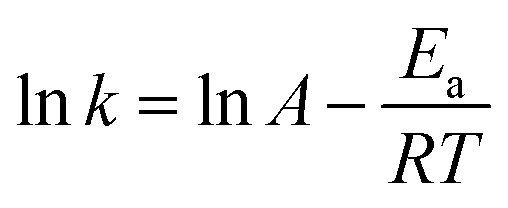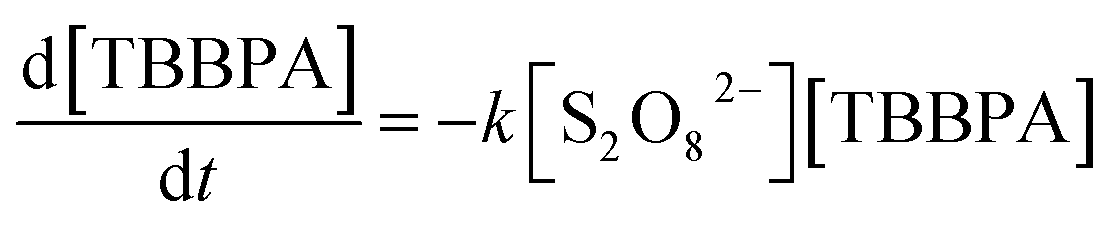DOI:
10.1039/C6RA02482C
(Paper)
RSC Adv., 2016,
6, 29718-29726
Degradation of tetrabromobisphenol A in heat activated persulfate oxidation process†
Received
27th January 2016
, Accepted 16th March 2016
First published on 17th March 2016
Abstract
Sulfate radicals (SO4˙−) generated by heat activated persulfate were employed to degrade brominated flame retardant tetrabromobisphenol A (TBBPA). Experimental results showed that the reaction rate was first order to the concentrations of both persulfate and TBBPA. The degradation of TBBPA was accelerated with increasing temperature. Radical scavenging tests using ethanol and t-butanol as probes revealed that SO4˙− was the dominant oxidizing species. The degradation efficiency was adversely affected by the presence of humic acid and HCO3−. No significant inhibition of TBBPA degradation was observed in the presence of Cl−. Seven brominated intermediates and products were identified by liquid chromatography-mass spectrometry. Based on this, debromination and electron transfer followed by β-scission were proposed to be the primary pathways of TBBPA degradation. The results of this study suggest that the heat activated persulfate process could be an effective approach to remove TBBPA in aqueous solution.
Introduction
Tetrabromobisphenol A (TBBPA) is a brominated flame retardant (BFR) widely applied in the manufacture of paper, textiles, plastics, electronics and upholstered furniture.1–3 Due to its intensive use and high persistency, TBBPA is found in various environmental matrices such as air conditioning filter dust,4 waters,5,6 sediments7 and organisms.6 For instance, TBBPA concentration up to 0.6 μg L−1 was detected in the water of the Mihe estuary in China.8 At a contaminated site in Israel, the concentration of TBBPA in the upper 20 cm of the soil layer was more than 50 mg kg−1. It was found that TBBPA could be transferred to deeper soil profile and contaminate groundwater.9 In addition, derivatives of TBBPA, such as TBBPA bis(allyl)ether (TBBPA BAE) and TBBPA bis(2,3-dibromopropyl)ether (TBBPA BDBPE) have also been detected in the environment.10 TBBPA is an endocrine disruptor and shows negative effects to mammals and human beings, inducing cytotoxicity, hepatotoxicity, nephrotoxicity and neurotoxicity.11–14 Therefore, proper treatment of TBBPA contaminated water and soil is required in order to decrease associated environment risks.
Activated persulfate oxidation process, as a promising in situ chemical oxidation (ISCO) technology, has been widely applied in field remediation in recent years. Persulfate is more stable than hydrogen peroxide, thereby allowing for great dispersion distance in subsurface. Freely diffusible sulfate radical (SO4˙−, E0 = 2.5–3.1 V)15 is believed to be the primary oxidizing species responsible for the degradation of pollutants in this process. SO4˙− is an electrophile that reacts with a wide range of contaminants through electron transfer mechanism with a second order rate constant ranging from 106 to 109 M−1 s−1.16 SO4˙− can be formed by activation of persulfate (PS) or peroxymonosulfate (PMS) via various approaches including heat, ultraviolet, transition metals, base, hydrogen peroxide, etc.15,17–19 Among these activation technologies, heat activation offers some advantages. For example, it can minimize the consumption of PS caused by pre-mixing with activator chemicals before injection.20 Increasing the activation temperature favors the decomposition of PS and SO4˙− formation, thus, accelerating the reaction to shorten the remediation time.15,18 Heat activation is also frequently employed to explore the reaction mechanisms between SO4˙− and various contaminants due to its simplicity and higher efficiency.21 In field practice, heat activation PS oxidation can be combined with in situ thermal remediation (ISTR)22 technology.
| |
 | (1) |
Oxidation of halogenated compounds by activated PS processes has been widely reported; examples include perfluorooctanoic acid,23 chlorinated solvents,18,24,25 polychlorinated biphenyls (PCBs),26,27 chlorinated pesticides,28,29 chlorophenols,30,31 and chloroanilines.19 Degradation of TBBPA in SO4˙−-based oxidation processes has also been documented. Guo et al. reported that UV/base/PS process could achieve complete degradation of TBBPA.32 Ding et al. revealed that TBBPA was efficiently degraded in sulfate radicals oxidation catalyzed by CuFeO4 magnetic nanoparticles.33 Fukushima et al. explored the oxidation of TBBPA by Fe-porphyrin/KHSO5 and found that TBBPA was rapidly removed from the solution in the absence and presence of humic acid (HA).34 Unfortunately, these studies were either conducted in extreme conditions (e.g., strong basic condition) or used specifically prepared catalysts that were uneconomic for practical application. As an alternative, heat activation of PS might be relatively cost effectiveness and technically feasible for TBBPA degradation. However, little is currently known about the degradation of TBBPA in heat activated PS process.
This study aims to evaluate the technical feasibility of degradation of TBBPA by heat activated PS in aqueous solution. Laboratory scale batch experiments were conducted to investigate the kinetics of TBBPA degradation. Factors affecting degradation, including temperature, PS dosage, chloride, carbonate and HA were assessed. Furthermore, we elucidate the transformation mechanisms and pathways for TBBPA reaction with SO4˙−.
Materials and methods
Reagents and materials
Tetrabromobisphenol A (99.0%), sodium persulfate (99.5%) were purchased from Aladdin (Shanghai, China). Humic acid (HA) was terrestrial derived and also obtained from Alladin. HPLC grade methanol and formic acid were purchased from Burdich & Jackson (Shanghai, China). Other reagents were at least of analytical grade and used as received. All stock solutions were prepared by dissolving the chemical agents in Milli-Q water (18 MΩ cm−1) prepared from a Millipore system (Bedford, USA) and used within 1 week.
Experimental setup
Unless otherwise specified, the degradation experiments were carried out in 33 mL screw-cap EPA vials with Teflon septa at desired temperatures controlled by a thermos-stated water bath. The solutions containing 5 mg L−1 TBBPA were pre-heated to the working temperature before appropriated volume of PS stock solution was spiked to initiate the reaction. Due to adding only a tiny amount of PS stock solution, changes in the temperature of the reaction solutions were negligible. No buffer was used to avoid potential reactions between the additives and SO4˙−. Initial pH was adjusted by 0.01 M H2SO4 or NaOH to desired value (8.0 ± 0.2). At preset reaction time, sample aliquots (1.0 mL) were withdrawn from the reaction vials, and chilled in an ice bath for 10 min to quench the reaction.20,35 Control experiments with PS absent were run concurrently under identical conditions. No degradation of TBBPA was found in the controls, indicating TBBPA was hydrolysis-resistant and heat stable.
To determine the effect of temperature on the TBBPA degradation, PS dose was fixed at 0.4 mM, five temperature regimes of aqueous solutions at 30, 40, 50, 60 and 70 °C were studied. In the remaining experiments, the reaction temperature was controlled at 50 °C. To determine the effect of PS dosages on the TBBPA degradation, initial PS concentrations of 0.2, 0.4, 0.6, 0.8 and 1.0 mM were used. To explore the effects of chloride or bicarbonate on the degradation of TBBPA, NaCl or NaHCO3 was spiked to the solutions before 0.4 mM PS was added to initiate the reaction. Four concentration levels, 2, 5, 10 and 100 mM, for each of the anions were tested. The effect of HA on TBBPA removal was studied in a similar manner. HA concentrations of 2.0, 4.0, 6.0, 8.0 and 10.0 mg L−1 (as TOC) were selected. In order to determine the radical species formed in the system, 10 mM tert-butyl alcohol (TBA) or ethanol (EtOH) was added into the reaction solution prior to the addition of 0.4 mM PS. Controls with the additives absent but other conditions identical were run concurrently. Two replicates were prepared for each condition. Results were averaged.
Analytical methods
TBBPA concentration was measured by high performance liquid chromatography (HPLC, Hitachi L-2000) equipped with a photo diode array detector. Analytical conditions were as follows: a C18 reverse phase column (5 μm × 250 nm × 4.6 mm) was employed for separation; an isocratic solvent consisted of 85% methanol and 15% water with a flow rate of 1.0 mL min−1 was used for elution; the injection volume was 50 μL; an L-2300 column oven was used to maintain the column temperature at 30 °C. TBBPA was quantified by multipoint standard calibration curves. Under these conditions, the retention time of TBBPA was 7.8 min and the quantification limit was 0.05 mg L−1.
TBBPA degradation products were separated and enriched by solid phase extraction (SPE) technique. An aqueous solution (50 mL) contained 5 mg L−1 TBBPA and 0.4 mM PS was allowed to react for 60 min at 50 °C before quenched in an ice bath. The mixture was concentrated by a Waters Oasis hydrophilic–lipophilic balance (HLB) SPE cartridge (WAT106202). Prior to extraction, the cartridge was activated by 5 mL methanol followed by 5 mL Milli-Q water. The quenched reaction solution was percolated through the cartridge at a flow rate of 5 mL min−1. The extracts were eluted with 2 mL methanol twice.
Reaction products were analyzed using an Agilent 6410 triple quadrupole mass spectrometer with electron-spray ionization (ESI) source. The ESI source was operated at negative mode and mass analyzer at full scan (m/z 50–1000). The operation conditions were as follows: flow rate of the delivery solvent consisting of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 methanol/water (v/v) was 0.2 mL min−1; capillary voltage 3.8 kV, fragmentor 120 V, drying gas flow 3 mL min−1 at 350 °C, sheath gas flow 10 mL min−1 at 350 °C and the nebulizer pressure 40 psi.
:50 methanol/water (v/v) was 0.2 mL min−1; capillary voltage 3.8 kV, fragmentor 120 V, drying gas flow 3 mL min−1 at 350 °C, sheath gas flow 10 mL min−1 at 350 °C and the nebulizer pressure 40 psi.
Results and discussions
Reaction kinetics
Preliminary tests were conducted to compare the performance of TBBPA degradation under different conditions, including PS absent and PS without heating. As shown in Fig. S1, ESI,† TBBPA was stable at the highest temperature (70 °C) used in this study. Fig. 1a depicts the degradation of TBBPA with respect to different activation temperature with the initial PS concentration fixed at 0.4 mM. Increased removal of TBBPA was found as the temperature increased from 40 to 70 °C, suggesting that PS could be effectively activated by heat. For example, at 40 °C, the TBBPA removal was only 5% after a reaction period of 120 min while complete removal of TBBPA was achieved in 20 min at 70 °C. The degradation of TBBPA can be well fitted with the pseudo-first-order kinetic model (R2 > 0.98) as shown in Fig. 1a. The reaction can be described by eqn (2),| |
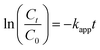 | (2) |
where Ct and C0 are the concentrations of TBBPA (mg L−1) at time t and zero, respectively; kapp(min−1) is the pseudo-first-order reaction rate constant. The rate constants were determined to be 0.0009, 0.0078, 0.0501 and 0.2217 min−1 at reaction temperatures of 40, 50, 60 and 70 °C, respectively. Increased degradation of TBBPA could be attributed to the enhanced generation of sulfate radicals at elevated temperatures.36,37 Compared with the fact that little TBBPA was removed in PS oxidation at ambient temperature, it can be concluded that the free radicals was the principal oxidizing species responsible for TBBPA removal rather than PS.
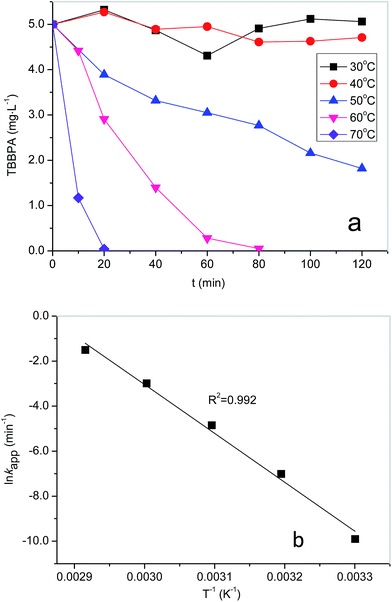 |
| | Fig. 1 (a) Effects of temperature on the removal of TBBPA in heat activated PS oxidation process, and (b) relationship between lnkapp and 1/T. Experimental conditions: [TBBPA] = 5 mg L−1, [S2O82−] = 0.4 mM, pH = 8.0. | |
The temperature dependency of kapp was evaluated using the Arrhenius equation (eqn (3)),
| |
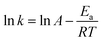 | (3) |
where
T (in Kelvin) is the absolute temperature,
A is the pre-exponential factor (unit of
k),
R is the gas constant and
Ea is the activation energy (kJ mol
−1).
Fig. 2b shows that ln
kapp decreased linearly with increasing of 1/
T, indicating their relationship fitting Arrhenius equation well. Activation energy
Ea was determined to be 180.5 kJ mol
−1 (
R2 = 0.99). The
Ea value of TBBPA is higher than those of other halogenated compounds in the heat activated PS oxidation system,
e.g., 108 kJ mol
−1 for trichloroethenes, 144 kJ mol
−1 for
cis-1,2-dichloroethene, and 141 kJ mol
−1 for
trans-1,2-dichloroethene.
18 This indicates TBBPA is more resistant to oxidation in heat activated PS process.
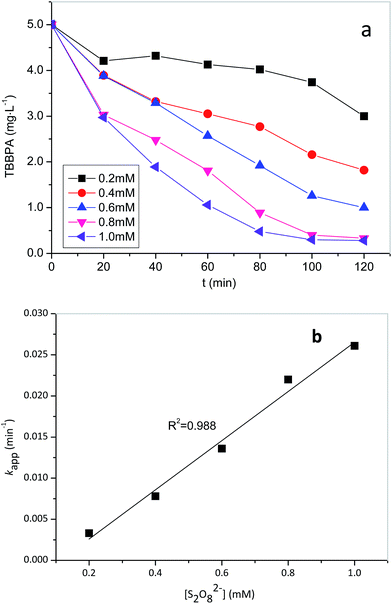 |
| | Fig. 2 (a) Effects of PS concentration on the removal of TBBPA in heat activated PS oxidation process, and (b) relationship between pseudo-first-order rate constant and PS concentration. Experimental conditions: [TBBPA] = 5 mg L−1, temperature = 50 °C, pH = 8.0. | |
The effect of PS dose on TBBPA removal was investigated at 50 °C. As shown in Fig. 2a, the degradation of TBBPA increased from 5% to 94% in 2 h as the initial concentration of PS increased from 0.1 to 1.0 mM. The removal of TBBPA at each PS dosage can be described with the pseudo-first-order kinetic model (R2 > 0.97). By fitting the experimental data to eqn (2), kapp values were determined to be 0.0033, 0.0078, 0.0136 and 0.0261 min−1 for the PS concentrations of 0.2, 0.4, 0.6 and 1.0 mM, respectively. It is evident that the values varied linearly with PS concentration (Fig. 2b), suggesting a first-order dependence of the reaction rate on PS concentration as described by eqn (4).
Substitution of (4) to (2), the reaction rate of TBBPA can be express as eqn (5), where k is the second-order reaction rate constant (M−1 min−1) and the value is 0.0289 × 103 M−1 min−1. According to this equation, it is evident that TBBPA oxidation by heat activated PS is first-order to the concentrations of both TBBPA and PS.
| |
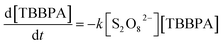 | (5) |
Determination of the radical species
Both SO4˙− and HO˙ could participate the degradation of contaminants in heat activated PS system. OH˙ can be formed by SO4˙− oxidation of hydroxyl ion at alkalinic conditions38 (eqn (6)).| | |
SO4˙− + OH− → SO42− + HO˙, k = (6.5 ± 1.0) × 107 M−1 s−1
| (6) |
In the present study, alcohols were used to probe the contributions of the two radicals. Ethanol (EtOH) having α-H reacts rapidly with both HO˙ and SO4˙− at second-order rate constants of 1.2–2.8 × 109 M−1 s−1 and 1.6–7.7 × 107 M−1 s−1, respectively. While tert-butyl alcohol (TBA) without α-H, the reaction rate constant with HO˙ was more than 3 orders of magnitude higher than that with SO4˙−, as shown in eqn (7) and (8).39
| | |
TBA + HO˙ → products, k = (3.8–7.6) × 108 M−1 s−1
| (7) |
| | |
TBA + SO4˙− → products, k = (4.0–9.1) × 105 M−1 s−1
| (8) |
Thus, EtOH can quench both SO4˙− and HO˙; while TBA preferentially quenches HO˙. According to the different selectivity of SO4˙− and HO˙ to different scavengers, the radical species responsible for the degradation of TBBPA in the reaction system can be determined. As shown in Fig. 3, degradation of TBBPA was significantly inhibited in the presence of excessive TBA. Even more suppression of TBBPA degradation was observed when EtOH was added. These results suggested that both HO˙ and SO4˙− played important roles in TBBPA degradation.
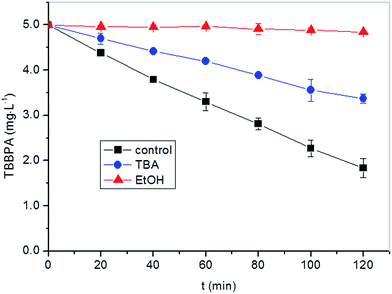 |
| | Fig. 3 Effects of EtOH and TBA as radical scavengers on the degradation of TBBPA in heat activated PS oxidation process. Experimental conditions: [TBBPA] = 5 mg L−1, [S2O82−] = 0.4 mM, [EtOH] = 10 mM, [TBA] = 10 mM, pH = 8.0. | |
Effects of natural water constituents
Natural organic matter (NOM) is ubiquitously present in natural waters. Since NOM is primarily comprised of humic substances,40 commercial HA was used to mimic the reactivity of NOM in this work. As shown in Fig. 4a, presence of NOM exhibited an inhibitive effect on the degradation of TBBPA and the extent of inhibition increased with the increasing of NOM concentration. For instance, TBBPA was degraded completely in 120 min without NOM while only 50% of TBBPA was eliminated in the presence of 10 mg L−1 NOM. NOM is known to be a sink of SO4˙− and OH˙ because certain functional groups, such as hydroxyl group, in NOM molecules are prone to react with the radicals.41,42 Therefore, the inhibition on TBBPA removal can be partly explained by competitive scavenging of radicals by NOM. Reactions between SO4˙− and NOM were previously explored and supported by density functional theory (DFT) calculation.43 On the other hand, NOM could severe as organic phase and organic pollutants trend to be imbedded in the NOM phase, making them less likely attacked by SO4˙− since the surrounding matrix (i.e., NOM) is itself reactive with SO4˙−.44 Therefore, higher concentration of NOM in aqueous leads to more pronounced inhibition on TBBPA removal.
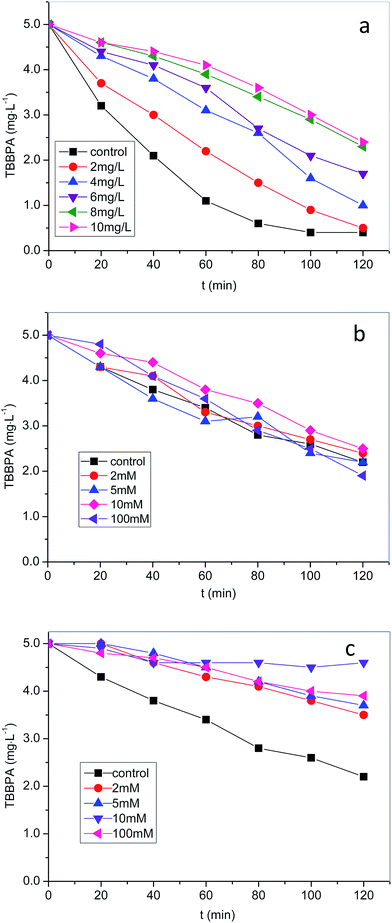 |
| | Fig. 4 Effects of (a) NOM, (b) Cl−, and (c) HCO3− on the degradation of TBBPA in heat activated PS oxidation process. Experimental conditions: [TBBPA] = 5 mg L−1, T = 50 °C, [S2O82−] = 1.0 mM for (a), [S2O82−] = 0.4 mM for (b) and (c), pH = 8.0. | |
Inorganic ions are also likely to affect the transformation of organics in persulfate oxidation processes.45 Thus, the effects of Cl− and HCO3− on the degradation of TBBPA were evaluated at 50 °C (Fig. 4b and c). As shown in Fig. 4b, the degradation of TBBPA was not influenced by Cl− at concentration up to 5 mM. It is known that Cl− can be oxidized by SO4˙− to form reactive chlorine species including chlorine radicals (Cl˙ and Cl2˙−) and free chlorine (Cl2 and HOCl).46,47 These reactive species are moderate oxidants and can selectively react with electron-rich compounds such as phenolic and anilinic compounds (eqn (9)–(14)).45,46 Thus, if the target contaminants fall into this category, the reactions with the reactive chlorine species could compensate the reduced SO4˙−. Otherwise, presence of Cl− would inhibit the degradation of contaminants. For instance, it was found that high concentration of Cl− (>100 mM) promoted sulfamethazine oxidation but suppressed the degradation of atrazine in heat activated oxidation process.37,48 In the present study, TBBPA was relatively electron-deficit due to the substitution of two bromine atoms on the aromatic ring. Its reaction with reactive chlorine species was presumably slow. Note that, the reaction between Cl− and SO4˙− is reversible with forward and backward reaction rates approximately same magnitude (eqn (9)). Since the formed Cl˙ was not readily consumed, the reaction would reach equilibrium quickly. Thus, we assume the consumption of SO4˙− by Cl− was insignificant in this study. As a result, only slightly inhibition of TBBPA degradation was observed. Similar finding was reported for atrazine by Ji et al.37
| | |
SO4˙− + Cl− ↔ SO42− + Cl˙, kf = 4.7 × 108 M−1 s−1; kr = 2.5 × 108 M−1 s−1
| (9) |
| | |
Cl˙ + Cl− ↔ Cl2˙−, k = 8 × 109 M−1 s−1
| (10) |
| | |
Cl2˙− + Cl2˙− → Cl2 + 2Cl−, k = 1.3 × 109 M−1 s−1
| (11) |
| | |
Cl2(aq) + H2O → HOCl + H+ + Cl−
| (12) |
| | |
R˙ + Cl2˙− → R–Cl + Cl−
| (13) |
| | |
R–H + HOCl → R–Cl + H2O
| (14) |
HCO3− is a dominant anion in natural waters. The impact of HCO3− on heat activated PS oxidation of TBBPA was investigated by varying the concentration of HCO3−. Compared to Cl−, HCO3− exhibited a significant inhibition on TBBPA removal as show in Fig. 4c. Increasing the concentration of HCO3− from 0 to 100 mM led to a decrease in TBBPA removal from nearly 60–5% in 120 min. Similar results were also observed in the degradation of carbamazepine and trichloroethylene in SR-AOPs.36,47 Similar to Cl−, HCO3− performed as radical scavenger to consume SO4˙−. However, the reaction between carbonate and SO4˙− is irreversible (eqn (15)). Thus, the scavenging effected of HCO3− would be significant even at relatively low concentration, which is distinct from that of Cl−. The formed HCO3˙/CO3˙− (eqn (15) and (16)) only react with electron-rich compounds. Degradation of TBBPA due to reactions with HCO3˙/CO3˙− was probably negligible.
| | |
SO4˙− + HCO3− → HCO3˙ + SO42−, k = (1.6 ± 0.2) × 106 M−1 s−1
| (15) |
| | |
HCO3˙ → CO3˙− + H+, pKa = 9.5 ± 0.2
| (16) |
Oxidation products and transformation pathways
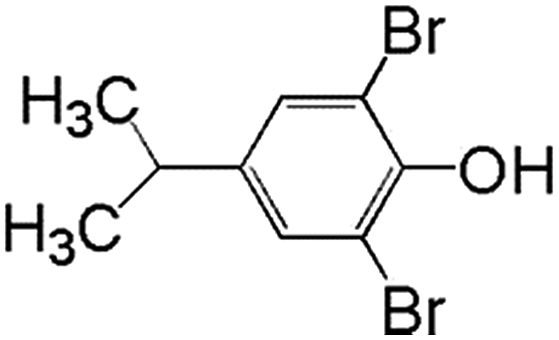
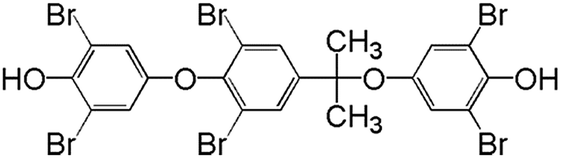
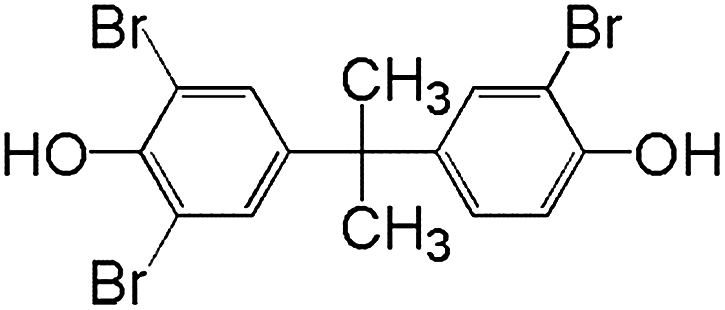
In order to understand the reaction pathways, intermediates and products of TBBPA degradation in heat activated PS oxidation process were separated and analyzed by MS equipped with an electrospray ionization (ESI) interface operated at negative mode. The chemical structures of intermediates and products were proposed on the basis of (i) their molecular weights (MWs); (ii) bromine isotope patterns (bromine has 2 natural isotopes (79Br and 81Br) with approximately equal abundance, a brominated compound should have an isotopic distribution pattern fitting to the binomial expansion of (1 + 1)n, where n is the number of bromine atoms in the molecule); and (iii) previously reported information on products formation during various oxidative processes involving TBBPA. Using this approach, a total of 7 brominated intermediates and/or products (P1–P7) were identified as shown in Fig. 5 and Table 1. The intermediates corresponding to ion clusters m/z 307/309/311 and m/z 289/291/293 contain 2 bromine atoms because of the isotopic distribution patterns approximately equal to 1:2:1. According to the MWs, peaks of m/z 307/309/311 and m/z 289/291/293 were assigned as 4-(2-hydroxyisopropyl)-2,6-dibromophenol and 4-isopropylene-2,6-dibromophenol, respectively. These intermediates were also observed by Ding et al. in heterogeneous catalytic PS degradation of TBBPA by CuFe2O4.33 Similarly, the peak of m/z 291/293/295 is assigned to be 4-isopryl-2,6-dibromopheol based on the MW and bromine isotope pattern. Time-dependent evolution profiles demonstrate that 4-isopropylene-2,6-dibromophenol and 4-isopryl-2,6-dibromopheol were formed sequentially (Fig. S2, ESI†). In addition, four products with relatively low signal response were found, suggesting that they were minor degradation products of TBBPA. Among of them, the products corresponding to ion clusters m/z 555/557/559/561/563, 803/805/807/809/811/813/815 and m/z 787/789/791/793/795/797/799 are likely coupling products of the degradation intermediates according to the isotopic abundance ratios and MWs. Another product corresponding to ion clusters m/z 461/463/465/467 was probably tribromobisphenol A (TriBBPA) resulting from debromination of TBBPA.
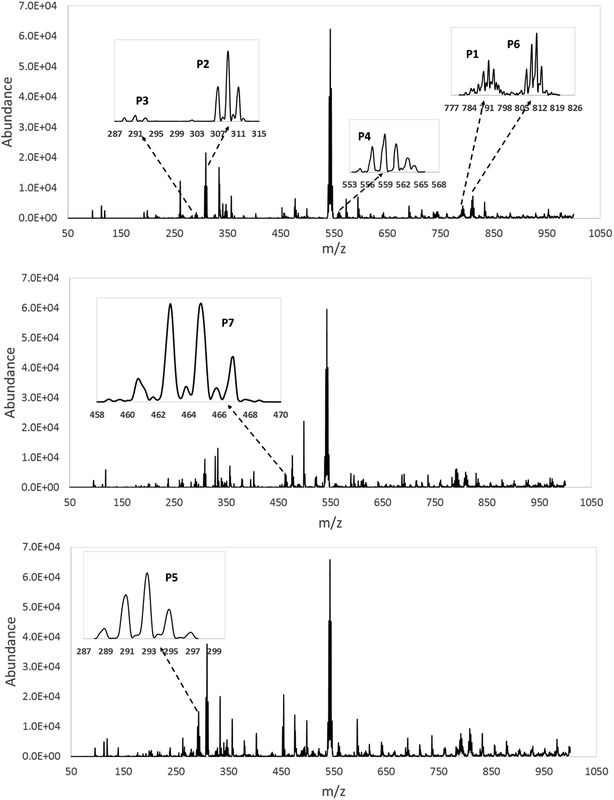 |
| | Fig. 5 Full scan mass spectra and detailed ion clusters of possible intermediates/products of TBBPA degradation. | |
Table 1 m/z data and proposed structure of intermediates of TBBPA
| # |
m/z (ion clusters) |
Isotopic distribution pattern |
Proposed structure |
| 1 |
787/789/791/793/795/797/799 |
1:6:15:20:15:6:1 |
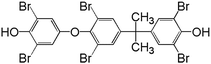 |
| 2 |
307/309/311 |
1:2:1 |
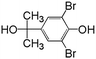 |
| 3 |
289/291/293 |
1:2:1 |
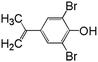 |
| 4 |
555/557/559/561/563 |
1:4:6:4:1 |
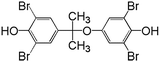 |
| 5 |
291/293/295 |
1:2:1 |
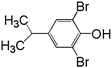 |
| 6 |
803/805/807/809/811/813/815 |
1:6:15:20:15:6:1 |
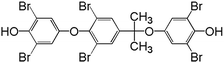 |
| 7 |
461/463/465/467 |
1:3:3:1 |
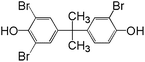 |
Based on the structures of intermediates and products as well as the oxidation mechanisms of SO4˙−, pathways for the oxidation of TBBPA in heat activated PS can be tentatively proposed. As showed in Fig. 6, degradation of TBBPA involved two different pathways (labeled as Route I and II), corresponding to two different sites for SO4˙− attack on TBBPA molecule. In Route I, one of the phenol moieties of TBBPA was attacked by SO4˙−, forming a phenoxy radical (R1) which could be stabilized by relocating the unpaired electron to the aromatic ring through resonance.49,50 TBBPA radical (R1) underwent β-scission (cleavage between the isopropyl group and one of the benzene rings) to release a carbocation (R2) and a radical (R3). Coupling of radicals R1 and R3 formed a dimeric product P1. The carbocation intermediate R2 was subjected to substitution or elimination reactions to give rise to more products. For instance, R2 could react with water or hydroxide to form P2 or eliminate a proton to form P3. Similar mechanism was also reported by Zhong et al., in the degradation of TBBPA by UV/Fenton and proposed that the primary reaction involved cleavage of the middle carbon atom.51 The intermediate R4 formed from R3 via reactions with water or hydroxide, as an important intermediate reported previously in the oxidative degradation of TBBPA, was not detected in the present work, possibly due to its rapid transformation. Condensation of R2 and R4 generated the product P4. However, P3 could be transformed to 4-isopropyl-2,6-dibromopheol (P5) through hydrogenation. Similar to the generation mechanism of P1, the dimeric product P6 could be possibly generated by reaction of P4 and R3. Route II was debromination. The C–Br bond of TBBPA was attacked by SO4˙− to form TriBBPA (P7). This pathway was also reported in oxidation of TBBPA by ozonation, photo-Fenton and UV/base/PS systems.32,52,53
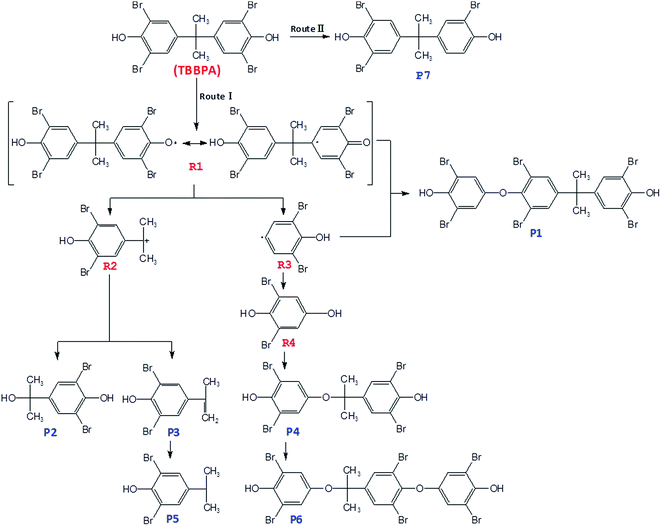 |
| | Fig. 6 Proposed transformation pathways of TBBPA in heat activated PS oxidation process. | |
Conclusions
Degradation of TBBPA was systematically examined in heat activated PS oxidation process in this study. It was demonstrated that increasing temperature and PS dosage significantly enhanced the degradation of TBBPA. TBBPA oxidation followed pseudo-first-order reaction kinetics. Radical scavenging test revealed that SO4˙− played a significant role in TBBPA removal. The effects of natural water constituents on TBBPA removal were investigated and results showed that HA and HCO3− exhibited inhibitive effect on the degradation of TBBPA; whereas Cl− showed a negligible effect. Based on the molecular structures of intermediates and products, two transformation pathways including debromination and electron transfer followed by β-scission, were proposed to be responsible for the degradation of TBBPA. Polybrominated products via radical coupling of the degradation intermediates were also found. The present study suggests that heat activated PS process is effective to eliminate TBBPA. Although this technique is relatively costly in operation, combination with in situ thermal remediation (ISTR) could make it an attractive option. However, it should be noted that debromination was minimal during TBBPA degradation and this could lead to a final mixture with similar or even higher toxicity than the original flame retardant. Thus, further toxicological tests are needed to thoroughly assess the toxic effects of the transformation products before this technology could be used in real practice.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (KYZ201407), China Postdoctoral Research Funds (2015M570454), and the priority Academic Program Development (PAPD) of Jiangsu Higher Education Institute.
References
- M. Alaee, P. Arias, A. Sjodin and A. Bergman, Environ. Int., 2003, 29, 683–689 CrossRef CAS PubMed.
- A. Covaci, S. Voorspoels, M. A.-E. Abdallah, T. Geens, S. Harrad and R. J. Law, J. Chromatogr. A, 2009, 1216, 346–363 CrossRef CAS PubMed.
- C. A. de Wit, Chemosphere, 2002, 46, 583–624 CrossRef CAS PubMed.
- H.-G. Ni and H. Zeng, Sci. Total Environ., 2013, 458, 15–19 CrossRef PubMed.
- S. Morris, C. R. Allchin, B. N. Zegers, J. J. H. Haftka, J. P. Boon, C. Belpaire, P. E. G. Leonards, S. P. J. Van Leeuwen and J. De Boer, Environ. Sci. Technol., 2004, 38, 5497–5504 CrossRef CAS PubMed.
- S. Yang, S. Wang, F. Wu, Z. Yan and H. Liu, Environ. Sci. Pollut. Res., 2012, 19, 4090–4096 CrossRef CAS PubMed.
- L. S. Morf, J. Tremp, R. Gloor, Y. Huber, M. Stengele and M. Zennegg, Environ. Sci. Technol., 2005, 39, 8691–8699 CrossRef CAS PubMed.
- H. Zhang and X. Yun, Chin. J. Environ. Eng., 2011, 5, 1077–1080 Search PubMed.
- S. Arnon, Z. Ronen, A. Yakirevich and E. Adar, Chemosphere, 2006, 62, 17–25 CrossRef CAS PubMed.
- G. Qu, A. Liu, W. Thanh, C. Zhang, J. Fu, M. Yu, J. Sun, N. Zhu, Z. Li, G. Wei, Y. Du, J. Shi, S. Liu and G. Jiang, Environ. Sci. Technol., 2013, 47, 4760–4767 CrossRef CAS PubMed.
- T. Colnot, S. Kacew and W. Dekant, Arch. Toxicol., 2014, 88, 553–573 CAS.
- F. Al-Mousa and F. Michelangeli, PLoS One, 2012, 7, e33059 CAS.
- S. Kim, J. Park, H.-J. Kim, J. J. Lee, G. Choi, S. Choi, S. Kim, S. Y. Kim, H.-B. Moon, S. Kim and K. Choi, PLoS One, 2015, 10, e0125213 Search PubMed.
- R. M. Evans, M. Scholze and A. Kortenkamp, PLoS One, 2012, 7, e43606 CAS.
- A. Tsitonaki, B. Petri, M. Crimi, H. Mosbaek, R. L. Siegrist and P. L. Bjerg, Crit. Rev. Environ. Sci. Technol., 2010, 40, 55–91 CrossRef CAS.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef CAS.
- M. Ahmad, A. L. Teel and R. J. Watts, Environ. Sci. Technol., 2013, 47, 5864–5871 CrossRef CAS PubMed.
- R. H. Waldemer, P. G. Tratnyek, R. L. Johnson and J. T. Nurmi, Environ. Sci. Technol., 2007, 41, 1010–1015 CrossRef CAS PubMed.
- I. Hussain, Y. Zhang, S. Huang and X. Du, Chem. Eng. J., 2012, 203, 269–276 CrossRef CAS.
- A. Ghauch, A. M. Tuqan and N. Kibbi, Chem. Eng. J., 2012, 197, 483–492 CrossRef CAS.
- M. G. Antoniou, A. A. de la Cruz and D. D. Dionysiou, Appl. Catal., B, 2010, 96, 290–298 CrossRef CAS.
- A. Tistonaki and B. Petri, Environ. Sci. Technol., 2010, 40, 55–91 CrossRef.
- Y.-C. Lee, S.-L. Lo, J. Kuo and Y.-L. Lin, Chem. Eng. J., 2012, 198, 27–32 CrossRef.
- C. J. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Chemosphere, 2004, 55, 1213–1223 CrossRef CAS PubMed.
- C. J. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Chemosphere, 2004, 55, 1225–1233 CrossRef CAS PubMed.
- G.-D. Fang, D. D. Dionysiou, D.-M. Zhou, Y. Wang, X.-D. Zhu, J.-X. Fan, L. Gang and Y.-J. Wang, Chemosphere, 2013, 90, 1573–1580 CrossRef CAS PubMed.
- G.-D. Fang, D. D. Dionysiou, Y. Wang, S. R. Al-Abed and D.-M. Zhou, J. Hazard. Mater., 2012, 227, 394–401 CrossRef PubMed.
- A. Romero, A. Santos, F. Vicente and C. Gonzalez, Chem. Eng. J., 2010, 162, 257–265 CrossRef CAS.
- C. Tan, N. Gao, Y. Deng, N. An and J. Deng, Chem. Eng. J., 2012, 203, 294–300 CrossRef CAS.
- G. P. Anipsitakis, D. D. Dionysiou and M. A. Gonzalez, Environ. Sci. Technol., 2006, 40, 1000–1007 CrossRef CAS PubMed.
- L. Xu, R. Yuan, Y. Guo, D. Xiao, Y. Cao, Z. Wang and J. Liu, Chem. Eng. J., 2013, 217, 169–173 CrossRef CAS.
- Y. Guo, J. Zhou, X. Lou, R. Liu, D. Xiao, C. Fang, Z. Wang and J. Liu, Chem. Eng. J., 2014, 254, 538–544 CrossRef CAS.
- Y. Ding, L. Zhu, N. Wang and H. Tang, Appl. Catal., B, 2013, 129, 153–162 CrossRef CAS.
- M. Fukushima, S. Shigematsu and S. Nagao, Environ. Chem. Lett., 2011, 9, 223–228 CrossRef CAS.
- A. Ghauch and A. M. Tuqan, Chem. Eng. J., 2012, 183, 162–171 CrossRef CAS.
- J. Deng, Y. Shao, N. Gao, Y. Deng, S. Zhou and X. Hu, Chem. Eng. J., 2013, 228, 765–771 CrossRef CAS.
- Y. Ji, C. Dong, D. Kong, J. Lu and Q. Zhou, Chem. Eng. J., 2015, 263, 45–54 CrossRef CAS.
- C. Liang, Z.-S. Wang and C. J. Bruell, Chemosphere, 2007, 66, 106–113 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- K. Ichihashi, K. Teranishi and A. Ichimura, Water Res., 1999, 33, 477–483 CrossRef CAS.
- P. Westerhoff, S. P. Mezyk, W. J. Cooper and D. Minakata, Environ. Sci. Technol., 2007, 41, 4640–4646 CrossRef CAS PubMed.
- C. Cuypers, T. Grotenhuis, K. G. J. Nierop, E. M. Franco, A. de Jager and W. Rulkens, Chemosphere, 2002, 48, 919–931 CrossRef CAS PubMed.
- P. M. David Gara, G. N. Bosio, M. C. Gonzalez, N. Russo, M. d. C. Michelini, R. Pis Diez and D. O. Martire, Photochem. Photobiol. Sci., 2009, 8, 992–997 Search PubMed.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- L. R. Bennedsen, J. Muff and E. G. Sogaard, Chemosphere, 2012, 86, 1092–1097 CrossRef CAS PubMed.
- Y. Yang, J. J. Pignatello, J. Ma and W. A. Mitch, Environ. Sci. Technol., 2014, 48, 2344–2351 CrossRef CAS PubMed.
- C. Liang, Z.-S. Wang and N. Mohanty, Sci. Total Environ., 2006, 370, 271–277 CrossRef CAS PubMed.
- Y. Fan, Y. Ji, D. Kong, J. Lu and Q. Zhou, J. Hazard. Mater., 2015, 300, 39–47 CrossRef CAS PubMed.
- K. Lin, W. Liu and J. Gan, Environ. Sci. Technol., 2009, 43, 4480–4486 CrossRef CAS PubMed.
- Y. Feng, L. M. Colosi, S. Gao, Q. Huang and L. Mao, Environ. Sci. Technol., 2013, 47, 1001–1008 CrossRef CAS PubMed.
- Y. Zhong, X. Liang, Y. Zhong, J. Zhu, S. Zhu, P. Yuan, H. He and J. Zhang, Water Res., 2012, 46, 4633–4644 CrossRef CAS PubMed.
- J. An, L. Zhu, N. Wang, Z. Song, Z. Yang, D. Du and H. Tang, Chem. Eng. J., 2013, 219, 225–237 CrossRef CAS.
- R. Qu, M. Feng, X. Wang, Q. Huang, J. Lu, L. Wang and Z. Wang, PLoS One, 2015, 10, e0139580 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02482c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.