
Functionalization of silica through thiol-yne radical chemistry: a catalytic system based on gold nanoparticles supported on amino-sulfide-branched silica†
Abstract
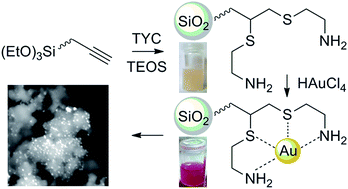
This work proposes a preparation route to heterogeneous catalysts based on gold nanoparticles (AuNPs) supported on chemically modified silica. More specifically, the latter is functionalized with amino-sulfide branches (Au-SiO2@AeThio) through a thiol-yne radical coupling performed between cysteamine hydrochloride and an alkynyl-substituted triethoxysilane, followed by co-condensation with tetraethoxysilane (TEOS). The target procedure, involving only a gold precursor without any need of additional reducing and/or stabilizing agents, is straightforward, controllable, reproducible, and particularly appealing from a “green” point of view. The supported AuNPs, with an average diameter of 10 nm, possess a remarkable catalytic activity (specific rate constants of the order of 10−2 s−1 mgcat−1) in the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) by sodium borohydride (NaBH4) in aqueous media. The higher performances with respect to previous literature work, along with the possibility of successfully recycling the catalyst, shows the developed materials as attractive functional platforms.
 Please wait while we load your content...
Please wait while we load your content...

