
AIE active triphenylamine–benzothiazole based motifs: ESIPT or ICT emission†
 *a
*a
Abstract
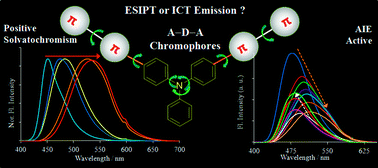
Two novel donor–π bridge–acceptor compounds containing excited state intramolecular proton transfer (ESIPT) and non-ESIPT units based on triphenylamine–benzothiazole were synthesized via Suzuki coupling reaction. Their photophysical properties were studied in the solid state as well as in solutions with solvents of different polarities. The fluorophores showed absorption in the UV region and emission in the visible region in both solutions and solid state. The optical properties of these compounds are highly dependent on solvent polarity. Significant positive solvatochromism (∼30 nm absorption and ∼80 nm emission red shift in polar solvents) were observed for both the compounds. Large Stokes shift (∼15 000 cm−1), polarity sensitive optical properties and very high quantum efficiencies (∼90%) in solvents and the solid state are the striking features of the synthesized compounds. Intramolecular charge transfer (ICT) characteristics of the compounds were supported experimentally and computationally. Halochromism and the intermolecular charge transfer phenomenon were used for investigation of ESIPT emission for compound 9 over ICT emission.
 Please wait while we load your content...
Please wait while we load your content...

