
Polyethylenimine incorporation into hydrogel nanomatrices for enhancing nanoparticle-assisted chemotherapy
Abstract
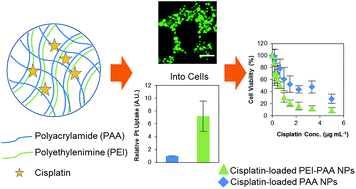
The efficacy of drug-loaded nanoparticles (NPs) for cancer therapy is related to (1) the cellular uptake of the NPs, (2) the ability to control drug release, and (3) the ability to deliver drugs to the cytosol, while evading the endosomal compartments. Here we present a NP carrier of cisplatin, made of poly(acrylamide-co-N-(3-aminopropyl)methacrylamide) (PAA) and a branched polyethylenimine (PEI), a source of primary, secondary, as well as tertiary amine groups. We found that (1) the cellular uptake of the NPs was highly enhanced by the PEI incorporation. However, surprisingly, this is not due to significant changes to the NP surface charge (zeta potential). Also, the PEI-incorporation into the PAA NPs resulted in (2) an accelerated release kinetics, (3) an ability to destabilize the endosomal/lysosomal membrane, and (4) a marginal increase in drug loading. The combination of the 4 aforementioned effects seems to account for the observed significant increase in the cytotoxicity of these cisplatin-loaded PEI-incorporated NPs, when applied to the 9L glioma cell line.
 Please wait while we load your content...
Please wait while we load your content...

