
Design, synthesis and biological evaluation of novel triazole-core reversal agents against P-glycoprotein-mediated multidrug resistance†
Abstract
We designed and synthesized a novel series of P-glycoprotein (P-gp)-mediated multidrug resistance (MDR) inhibitors bearing a triazolphenethyl–tetrahydroisoquinoline scaffold through click chemistry. Then those synthesized compounds were tested on doxorubicin-resistant erythroleukemia K562/A02 cells in a series of experiments. The result is that most of the synthesized compounds exhibit more active MDR reversal activity than verapamil (VRP). However, among them, compound 11 exhibits less cytotoxicity (IC50s > 100 μM), but more potency than the known P-gp inhibitors WK-X-34 and VRP on increasing anticancer drug accumulation in K562/A02 cells. Moreover, this compound can not only maintain a longer chemo-sensitizing effect (>24 h) than VRP (<6 h) with reversibility, but also significantly reverses MDR in a dose-dependent manner. Therefore, considering the low intrinsic cytotoxicity but strong reversal activity in vitro, compound 11 may be a promising lead with a potent and safe resistance modulator in combinational cancer chemotherapy.
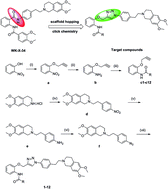
- This article is part of the themed collection: A Decade of Progress in Click Reactions Based on CuAAC
 Please wait while we load your content...
Please wait while we load your content...

