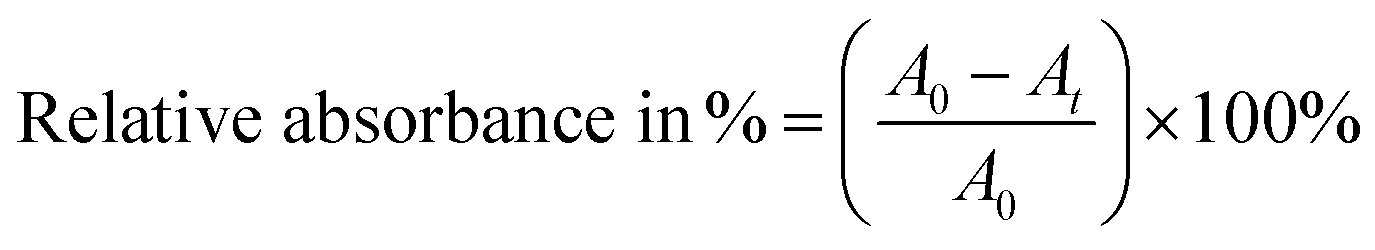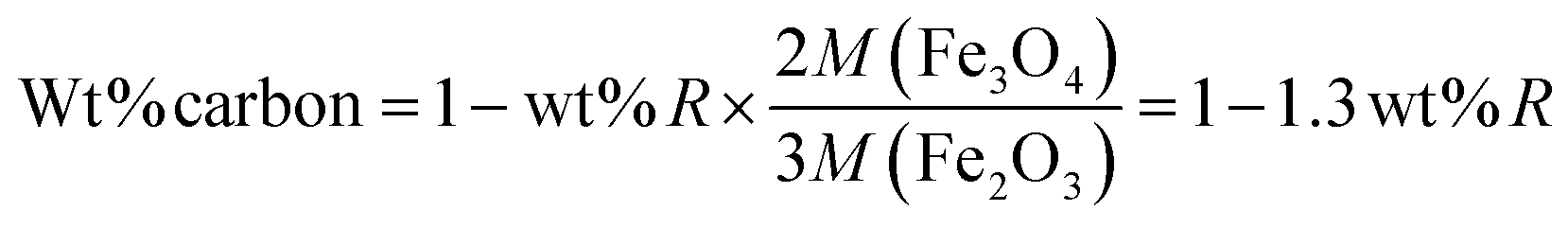DOI:
10.1039/C6RA02359B
(Paper)
RSC Adv., 2016,
6, 34297-34311
Stable and magnetically reusable nanoporous magnetite micro/nanospheres for rapid extraction of carcinogenic contaminants from water†
Received
26th January 2016
, Accepted 29th March 2016
First published on 31st March 2016
Abstract
We report the growth of porous magnetite microspheres via a template-free hydrothermal route at relatively low temperatures. The growth parameters including the concentration of reactant, reaction temperature and reaction time were examined for the formation of nanopores in the microspheres. Ascorbic acid played a crucial role in the formation of nanoporous microspheres, and also acts as a reducing agent, resulting from Fe3+ conversion into Fe2+ iron during the hydrothermal process. The resulting samples were used to remove acid orange 7 (AO7) and Cr(VI) from water at room temperature via adsorption. The maximum AO7 adsorption capacity calculated from the Langmuir–Freundlich model was found to be 69.13 mg g−1 for an optimum porous structure, which is significantly greater than that of structures formed without ascorbic acid (i.e., 4.61 mg g−1). Simultaneously, the Cr(VI) removal performance of the porous structure (i.e., 96.98 mg g−1) was found to be superior than that of pristine structure (i.e., 10.67 mg g−1). The enhanced removal activity was attributed to the existence of a large number of nanopores, resulting in a large specific surface area of 87.36 m2 g−1. Recycling tests were carried using a magnetic field to separate the powder samples from water, and the samples were found to be stable for up to three cycles. These results show that these porous microspheres have potential applications in cost-effective wastewater treatment.
Introduction
Recently, removal of heavy metal ions and toxic organic dyes from wastewater has become an important issue, because of the carcinogenic properties of these pollutants and their effects on human health and environment.1 Various techniques including advanced oxidation, coagulation, adsorption, reverse osmosis, and ion exchange have been used to remove these hazardous pollutants from polluted water.2,3 Of these, the adsorption technique is the most convenient and promising approach using different adsorbents, and is applicable to remove a wide range of pollutants. It has been used widely due to its simplicity, that its environmentally benign and produces less byproducts.4,5 Recently, diverse absorbents such as nickel oxide (NiO), activated carbon, metal–organic frameworks (MOFs), and porous structures with a large specific surface area, were employed to adsorb various pollutants.6–8 Nevertheless, these structures suffer poor performance and they are hard to separate from aqueous solution, which increases the total device cost. In addition, high surface free energy of small particles leads to agglomeration. Alternatively, porous nanostructures with strong magnetic properties have been broadly studied due to their high surface area and quick separation ability.9–11 In particular, nanoporous structures are suitable for efficient and advanced devices due to the large number of active centers, the easy recovery and long-term stability.12 The widespread use of nanostructures based on metal oxides such as TiO2, ZnO and iron oxide (FeOOH/Fe2O3/Fe3O4), as well as carbonaceous materials, has been reported for applications as absorbents to remove contaminants.13–15
Magnetite (Fe3O4) is an important oxide, and is a promising material for efficient and rapid separation of contaminants from aqueous solutions using a simple magnetic process. It has attracted much attention due to its magnetic and electrochemical properties, environmentally benign nature, natural abundance, large electrical conductivity, and high storage capacity in batteries.16,17 Furthermore, it can form various nanostructures, including flower-like, hollow spheres, chestnut-like, nanoplates, triangular nanosheets, nanorods and monodispersed spheres, which have potential applications in wastewater treatment, lithium batteries, photocatalysis, gas sensors, photoelectrochemical cells, pigments and hydrogen production.18–21 Most of these structures were fabricated using etching, self-assembly, hard/soft template, or anodization processes.22–24 Among these, solution-based self-assembly is the most viable method of producing porous three-dimensional (3D) structures, because it is a simple process, that requires relatively low temperatures and low cost, whereby the presence of foreign reactants within the growth solution can determine the final shape and size of the structures. For these reasons, many researchers have used solution processes to grow magnetic nanostructures.25–28 For example, Li et al. fabricated porous flower-like nanostructures via decomposition of alkoxide precursors in the presence of ethylene glycol, urea and iron chloride,29 and Xiong et al. grew single-crystal magnetite microspheres using a similar route, with the addition of sodium acetate and polyvinylpyrrolidone for applications in lithium-ion batteries.30 Mou et al. synthesized Chestnut-like hierarchical nanostructures using a solvothermal reaction for applications in the removal of arsenic, with rapid adsorption and quick separation from solution.31 Fan et al. reported a large specific surface area of 93.7 m2 g−1, and efficient adsorption of organic dyes and graphene with carbon-coated magnetite nanoparticle hybrids, which were fabricated using a one-pot solvothermal method.32 Wang et al. demonstrated a chemical template using a sonication approach to form composite orange-like magnetite and polypyrrole, which exhibited superior properties as a regenerated Cr(VI) adsorbent.33 Ma et al. reported excellent lithium storage properties of hollow magnetite spheres organized with ultrathin nanosheets using a facile solvothermal approach.34 However, fabrication of magnetite porous 3D structures has not yet been demonstrated using affordable methods for applications in wastewater management. Moreover, porous magnetite structures have not been extensively investigated for multi-pollutant removal.
Herein, we described a one-step and template-free approach to grow porous magnetite microspheres with a large number of nanopores using a relatively low-temperature process. We used ascorbic acid as structure directing agent to control porosity and morphology. Further, the influence of ascorbic acid, reaction temperature and time period to investigate the growth mechanism of porous structures. The resulting nanostructures grown at different growth conditions were employed to remove the carcinogenic pollutants acid orange 7 and Cr(VI) from water. Finally, magnetic separability and reusability over three cycle tests were demonstrated to show the stability of grown porous structures.
Experimental section
Growth of iron oxide nanostructures without and with AA
Nanoporous magnetite microspheres were grown via a hydrothermal route at relatively low temperature. Initially, 2.10 g of hexamethylenetetramine (HMTA, 0.5 M) was added into 30 mL of water at room temperature with stirring until a clear solution was obtained. Subsequently, 4.05 g of iron chloride (FeCl3·6H2O, 0.5 M) was added with stirring in the above solution. After this, 30 mL of absolute ethanol was poured into same solution. Then, various quantities of ascorbic acid (AA) powder (i.e., 1.0, 1.5 and 2.0 g) were then added. After stirring for 30 min, a homogeneous dark brown solution was formed, which indicates the reduction of Fe3+ to Fe2+. The homogeneous solution was then transferred into a Teflon-lined stainless steel autoclave, and allowed to react at 180 °C for 5 h. Following completion of reaction, the autoclave was allowed to cool to room temperature in air. A brown precipitate formed at the bottom of the Teflon-lined vessel, which was collected via filtration and washed several times with distilled water. The products were labeled as follows: without AA was labeled AA0/180, and with 1.0, 1.5 and 2.0 g of AA were labeled AA1.0/180, AA1.5/180 and AA2.0/180, respectively. To investigate the growth process of the porous spheres, we performed hydrothermal experiments at temperatures of 120, 140, 160 and 200 °C for 5 h with 1.5 g of AA; the products of these reactions are labeled AA1.5/120, AA1.5/140, AA1.5/160 and AA1.5/200, respectively. Finally, we varied the reaction time from 2 to 30 h, while maintaining the other parameters constant. The products reacted for 2, 10 and 30 h were labeled AA1.5/2 h, AA1.5/10 h and AA1.5/30 h, respectively. The collected powder samples were used directly for measurements and characterizations.
Removal of Cr(VI) and AO7
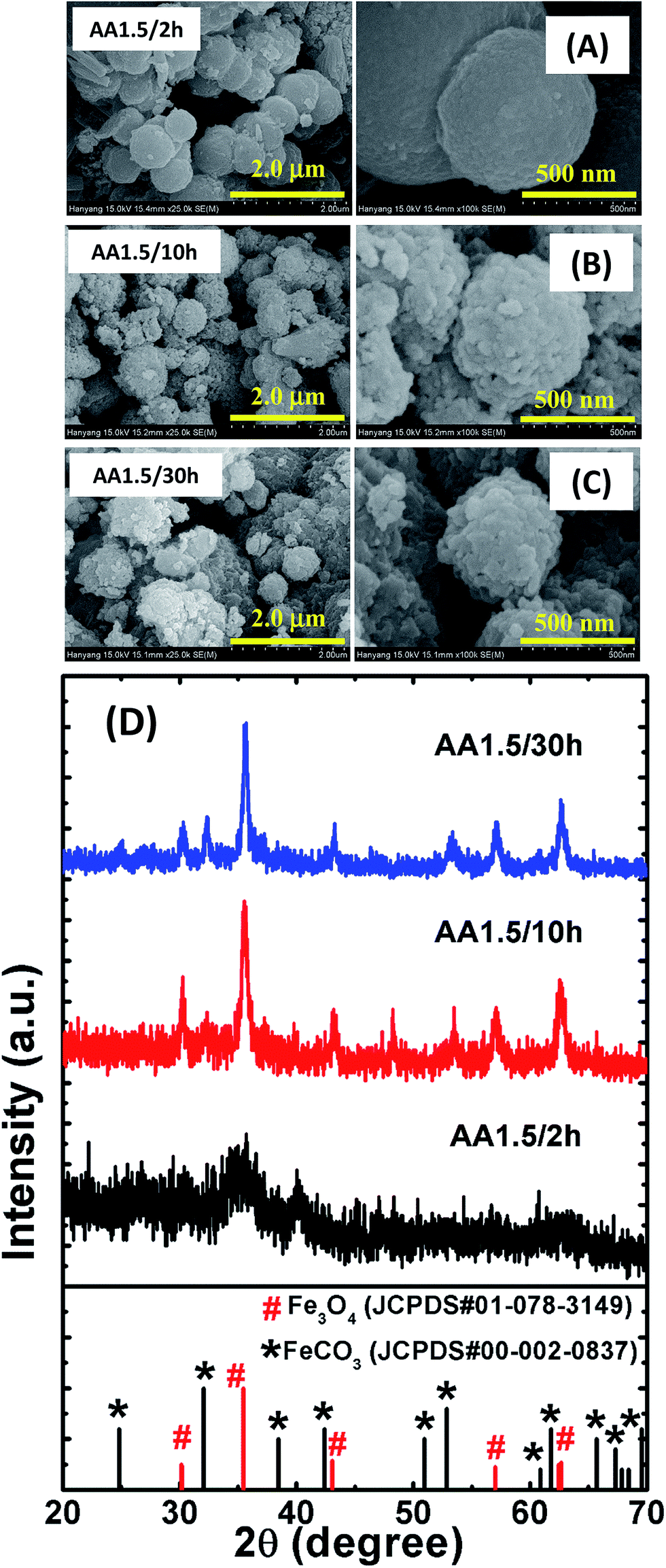
The adsorption capacity of the nanostructures was evaluated using a batch method. Stock solutions of Cr(VI) (K2Cr2O7, 200 mg) and AO7 (30 mg) were prepared by dissolving powder samples in 1000 mL of distilled water. The initial and final concentrations of Cr(VI) and AO7 were monitored using an ultraviolet-visible (UV-vis) spectrophotometer at wavelengths of 372 nm and 483 nm, respectively. To determine the equilibrium adsorption kinetics, 50 mg of catalyst powder were mixed in 50 mL of Cr(VI)/AO7 solution in a conical flask, and the resulting suspension was kept in a shaker and stirred at a speed of 200 rpm. Following specific time intervals, 5 mL of the solution were collected and centrifuged at 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min to separate the catalysts particles from the solution. A clear solution was obtained and used for the absorbance measurements directly. The change in absorbance was used to calculate the remaining concentration of contaminants. For the further studies on the adsorption kinetics, the pH of the Cr(VI) solution was adjusted from 3 to 11 using 0.5 M HCl and 0.5 M NaOH solutions. Using the absorbance data, we calculated relative absorbance of Cr(VI)/AO7 (in %) using the following expression:
000 rpm for 10 min to separate the catalysts particles from the solution. A clear solution was obtained and used for the absorbance measurements directly. The change in absorbance was used to calculate the remaining concentration of contaminants. For the further studies on the adsorption kinetics, the pH of the Cr(VI) solution was adjusted from 3 to 11 using 0.5 M HCl and 0.5 M NaOH solutions. Using the absorbance data, we calculated relative absorbance of Cr(VI)/AO7 (in %) using the following expression:| |
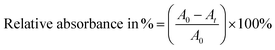 | (1) |
To confirm the complete removal of AO7 dye molecules, we measured the total organic carbon (TOC) of remaining solution using a Shimadzu TOC analyzer (TOC-VCSH, Japan).
Instrumentation
The powder samples collected following the hydrothermal treatment were analyzed using various techniques. The phase and crystalline orientation of the iron oxide microspheres were investigated using X-ray powder diffraction (XRD) (Rigaku D/MAX-2500/PC) using a Cu Kα radiation source. The changes in surface morphology with respect to the growth temperature, amount of ascorbic acid and reaction time were studied using field-emission scanning electron microscopy (FESEM) (Hitachi s4800, at 15 kV) and high-resolution transmission electron microscopy (HRTEM) (JEOL 2100 at 200 kV). Fourier transform infrared spectroscopy (FTIR) was used to determine the presence of various organic bonds and investigate the phase transformation of the iron oxide. The powders were mixed with KBr and pressed into pellets. FTIR spectra were recorded in the range 4000–400 cm−1 using a Bruker IFS 66/S. The Brunauer–Emmett–Teller (BET) specific surface area SBET was measured based on multipoint nitrogen adsorption–desorption isotherms using a Quantachrome analyzer (AS1, USA), and the Barrett–Joyner–Halenda (BJH) method was used to obtain the corresponding pore size distribution. The chemical composition of the nanostructures was analyzed using X-ray photoelectron spectroscopy (XPS) (Sigma Probe; Thermo-Scientific, UK). Magnetic characterization of the powder samples was carried out using a vibrating sample magnetometer (VSM) (Lake Shore, 7410 model, USA) at room temperature with a maximum applied magnetic field of 10 kOe.
Results and discussion
Quantity of ascorbic acid
Fig. 1 shows FESEM images of the hydrothermally grown iron oxide nanostructures with and without AA. The samples grown without AA were rhombohedral with an average diameter of 80 nm, as shown in Fig. 1A. Most of the particles were rhombohedral and uniformly distributed over the entire surface of the sample. To increase the surface area, we explored the growth of iron oxide nanostructures with AA to control the porosity and geometry. Initially, the quantity of AA was varied through 1, 1.5 and 2 g to optimize the growth of the mesoporous spherical iron oxide nanostructures, while maintaining equimolar (i.e., 0.5 M) quantities of FeCl3 and HMTA. The temperature of the reaction was 180 °C and the reaction time was 5 h. The morphology of the nanostructures changed from irregular particles to porous structures, which became larger as the AA concentration increased. With 1 g of AA, micron-sized particles with a random shape distribution grew throughout the entire area, as shown in Fig. 1B. The magnified image shows the rough surface of the micron-sized particles, which formed due to agglomeration of smaller particles. As we increased the quantity of AA to 1.5 g (i.e., the AA1.5/180 sample), the polydispersed microparticles became porous micro/nanospheres, as shown in Fig. 1C. The surface of these spheres exhibited a large number of pores, formed from number of self-assembled particles. A further increase of the quantity of AA to 2 g resulted in near perfect microspheres, with a smoother surface, as shown in Fig. 1D.
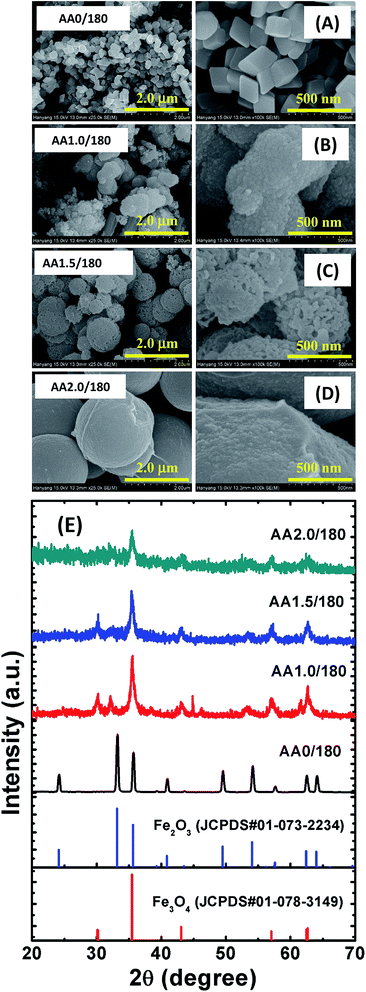 |
| | Fig. 1 FESEM images of iron oxide nanostructures grown with various quantities of ascorbic acid. (A) Without AA, (B) 1.0 g of AA, (C) 1.5 g of AA and (D) 2.0 g of AA. The left-hand side images have a magnification of 25000× and the right-hand side images have a magnification of 100000×. (E) XRD patterns of those samples. XRD patterns of magnetite and hematite from standard JCPDS data are also shown for reference. | |
Fig. 1E shows the XRD patterns of the nanostructures. A diffraction pattern of iron oxide grown in the absence of AA is provided for comparison, as well as standard Joint Committee on Powder Diffraction Standards (JCPDS) data card for Fe2O3 and Fe3O4. The powder samples collected without and with AA from hydrothermal process exhibited different phases. With the AA0/180 sample, strong and sharp diffraction peaks were observed at 2θ = 24.21°, 33.21°, 35.70°, 40.93°, 49.53°, 54.12°, 62.51° and 64.06°, which can be indexed as the crystalline rhombohedral phase of hematite (Fe2O3), which matched with the standard JCPDS data (card no. 01-087-1164). The nanostructures grown with AA showed magnetite (Fe3O4) phases of iron oxide, and the XRD patterns are consistent with the standard JCPDS data values (card no. 01-078-3149). For the AA1.0/180 sample, the diffraction peaks were strong and sharp, which is indicative of more crystalline phase of the Fe3O4 nanostructures. For the AA1.5/180 and AA2.0/180 samples, the intensity of the diffraction peaks decreased, illustrating the crucial role of AA in the crystallization of the Fe3O4 phase. Moreover, the structural phase transformation from hematite to magnetite can be attributed to the reducing properties of AA; i.e., it reduces Fe3+ to Fe2+.35 These experiments show that the quantity of AA was significant in the formation of the porous micro/nanospheres. Moreover, the nanostructures grown with 1.5 g of AA exhibited a large number of pores and were relatively crystalline. This may be expected to provide conduits for the adsorption of contaminant of molecules, as well as magnetization to improve the removal efficiency and reusability for applications in water purification.
Effects of the reaction temperature
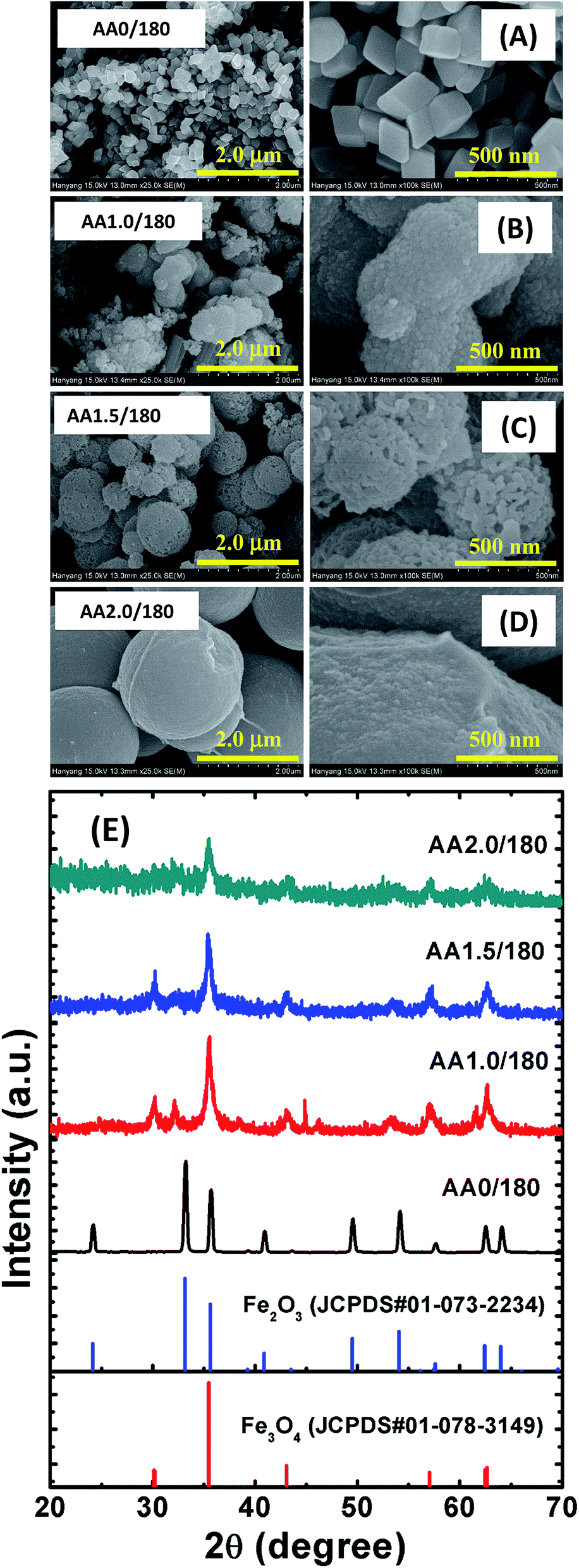
We grew porous nanostructures via a chemical route in which the parameters including, the reaction temperature and time, affected the surface morphology. Fig. 2A–E show representative FESEM images of the iron oxide nanostructures obtained via reactions at temperatures in the range 120–200 °C, while the other conditions were identical to those used for the synthesis of the AA1.5/180 sample. The temperature significantly affected the pore size distribution. Also note that the average dimensions of the spherical structures remained almost constant regardless of the temperature. Furthermore, the obtained spherical structures had different surface roughness: the spherical structure grown at low temperature (AA1.5/120) had smooth surfaces and no pores, which may have resulted from the low reaction temperature leading to slow evaporation of AA during the formation of the spheres (see Fig. 2A). Interestingly, the spheres grown at low temperature were amorphous. As we increased the reaction temperature, nanopores began to form inside the spheres, and the surface became rougher, demonstrating that higher temperatures assisted with the growth of the porous spheres (i.e., samples AA1.5/140 and AA1.5/160, as shown in Fig. 2B and C). In particular, sample AA1.5/180 (grown at 180 °C) exhibited nanopores due to the aggregation of particles, as described above. Importantly, the structure of the Fe3O4 phase changed from amorphous to crystalline the temperature increased. The low-magnification images show a uniform distribution of porous spheres throughout the entire area of sample (see Fig. 2D). The nanostructures obtained at 200 °C exhibited randomly shaped particles; however, the high reaction temperature led to the growth of nanostructures with many different shapes (i.e., AA1.5/200, as shown in Fig. 2E), and where the number of pores was smaller than with sample AA1.5/180. It follows that the high reaction temperature induced rapid growth of particles, leading to compact spheres. Furthermore, because of the high temperature, the surface morphology transformed from spherical to randomly shaped particles.
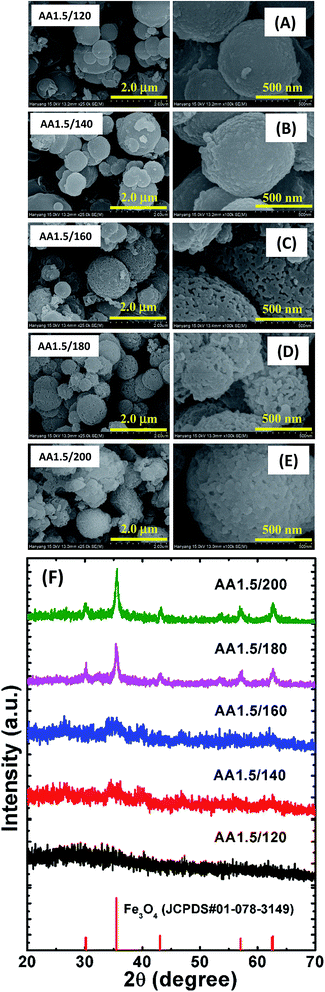 |
| | Fig. 2 FESEM images of samples with reaction temperatures of (A) 120 °C, (B) 140 °C, (C) 160 °C, (D) 180 °C and (E) 200 °C. The left-hand images have a magnification of 25000× and the right-hand images have a magnification of 100000×. (F) XRD patterns of those samples. An XRD pattern of magnetite from standard JCPDS data is also shown for reference. | |
These structures were further analyzed using XRD, as shown in Fig. 2F. Standard JCPDS data are also shown for reference. A crystalline magnetite phase was evident in sample AA1.5/180, and was more clearly evident in the sample grown at 200 °C. The absence of diffraction peaks corresponding to Fe3O4 in samples AA1.5/120, AA1.5/140 and AA1.5/160 indicates that these nanostructures were amorphous. As we increased the reaction temperature from 120 to 200 °C while maintaining all other conditions constant, the peaks related to Fe3O4 phase (i.e., 2θ = 30.23°, 35.55°, 43.21°, 57.57°, 62.34° and 62.71°) began to increase in magnitude, and were strongest for sample AA1.5/200. This shows that high reaction temperatures of 180 or 200 °C were required for the growth of crystalline Fe3O4. Moreover, the broad diffraction peaks indicate the nanocrystalline nature of the Fe3O4 structures. From these results, it is clear that the reaction temperature was significant in the geometry and crystallinity of the porous micro/nanospheres.
Reaction time
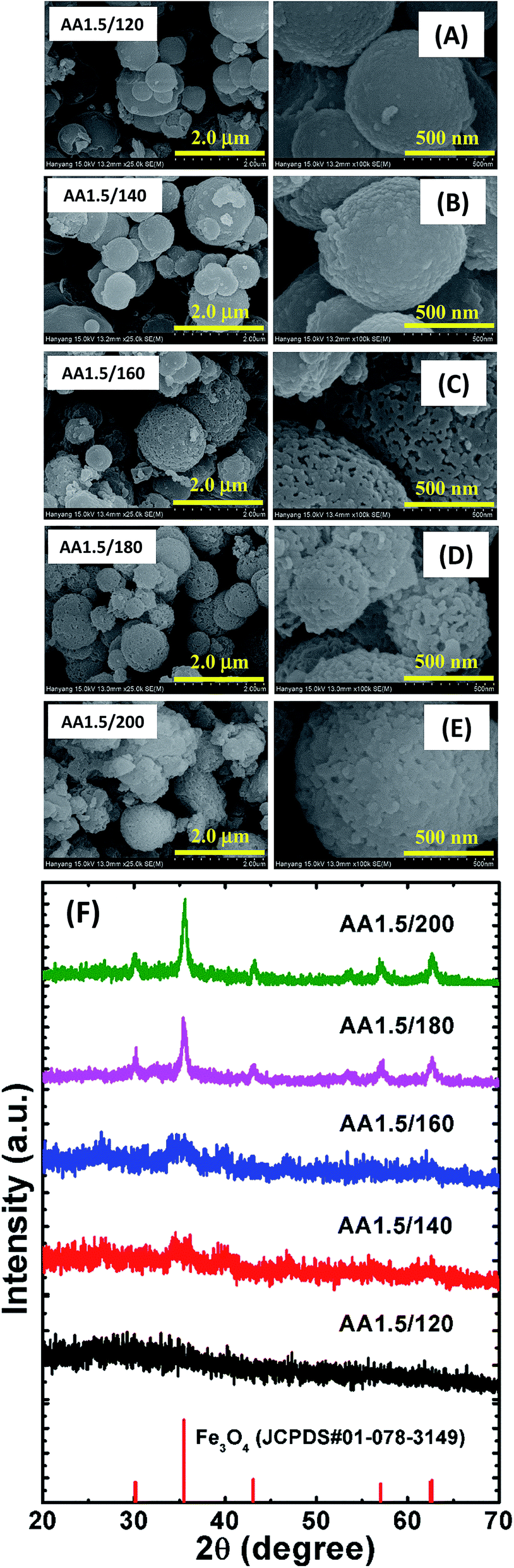
It is known that the nucleation and growth processes of nanostructures are dependent on the reaction time. To further investigate the growth process of the micro/nanospheres, we grew samples using different reaction times (i.e., samples AA1.5/2 h, AA1.5/10 h and AA1.5/30 h) while maintaining all other parameters constant, as shown in Fig. 3. Magnified images are shown on the right-hand side of Fig. 3A–C. The morphology of the micro/nanospheres was found to be sensitive to the reaction time. In the early stage of growth (i.e., sample AA1.5/2 h), smooth spheres formed with diameters in the range 0.5–2 μm, and the morphology was similar to that of the sample grown at low temperature. As demonstrated in the low-magnification image shown in the left-hand side image of Fig. 3A, the sample included some irregular particles in addition to the spherical nanoparticles. This indicates that the short reaction time period was not sufficient to form porous spheres. When we increased the reaction time to 5 h, almost all particles transformed into microspheres and were mono-dispersed with a diameter in the range 0.5–1 μm as discussed above. Sample AA1.5/10 h exhibited larger spheres, with diameters in the range 0.7–2 μm, as well as a rougher surface than the samples with shorter reaction times (see Fig. 3B). Careful observations reveal that most of the spheres were compact without nanopores. This result shows that a prolonged reaction time significantly affected the surface morphology and porosity of the microspheres. With a further increase in the reaction time to 30 h (i.e., sample AA1.5/30 h), the size of spheres remained unchanged, which illustrates that increasing the reaction time beyond 10 h had little effect on the surface morphology (see Fig. 3C). Moreover, the size distribution and porosity of the spheres was similar with that of sample AA1.5/10 h. The XRD patterns show that the reaction time was significant role in the formation of the crystalline Fe3O4 phase (see Fig. 3D). With a reaction time of 2 h, the nanostructures were almost amorphous, with only a peak at 2θ = 35.55°, and a further increase in the reaction time led to the formation of a crystalline phase (i.e., sample AA1.5/5 h). With longer reaction times of 10 and 30 h, strong and relatively sharp peaks were observed in the XRD patterns, indicating the formation of crystalline Fe3O4. It is known that the degree of nucleation increases with time, allowing the formation of a stable magnetite phase.27 Simultaneously, two additional peaks at 24.8° and 32.32° match with the ones for the iron carbonate phase (JCPDS#00-002-0837). This could be resulted from the conversion of iron into iron carbonate (FeCO3) due to small amount of Fe2+ ions, coordinating with AA molecules after specific reaction time period. The crystallinity of the samples formed with a reaction time of 30 h was similar to that of the sample formed with a reaction time of 10 h, indicating saturation in the growth of the microspheres.
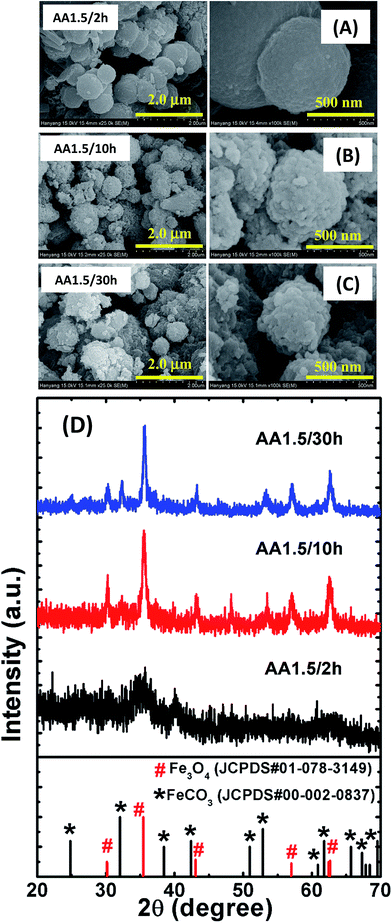 |
| | Fig. 3 FESEM images of samples formed with times of (A) 2 h, (B) 10 h and (C) 30 h. The left-side images have a magnification of 25000× and the right-hand images have a magnification of 100000×. (D) XRD patterns of those samples. An XRD pattern of magnetite (red color vertical lines with sphere symbol) and iron carbonate (black color vertical lines with square symbol) from standard JCPDS data are shown for reference. | |
TEM analysis
To characterize the detailed structural properties of the nanopores, we analyzed selected samples using TEM. Fig. 4A–G show representative TEM images of the samples formed with different reaction temperatures (i.e., AA1.5/120, AA1.5/140, AA1.5/160, AA1.5/180 and AA1.5/200) as well as different reaction times (i.e., AA1.5/2 h and AA1.5/10 h), with a constant quantity of AA of 1.5 g and an equimolar precursor concentration of 0.5 M. We can see that the AA1.5/120 and AA1.5/140 samples exhibited similar size distributions, with very dense spheres with an average diameter of 1 μm (see Fig. 4A and B). The magnified TEM image shows that there was a small change in the edges of the microspheres between the sample formed at 140 °C and that formed at 160 °C (see the high-magnification image in Fig. 4B). By increasing the reaction temperature to 160 °C, we obtained a monodisperse spherical morphology with thinner edges than for the samples grown at 140 °C (see Fig. 4C). When the reaction temperature was further increased to 180 °C, the surface morphology changed drastically, and its porosity also differed as compared to former structures (see Fig. 2D). In particular, the edges of the spheres were thinner compared with the sample grown at 180 °C (see the magnified image in Fig. 4D). There were fewer pores in the sample grown at 200 °C, indicating a porous structure of the microspheres (see the high-magnification image in Fig. 4E); however, at a reaction temperature of 200 °C, the spherical form became distorted agglomerated irregular particles formed (see Fig. 4E). The edges of the spheres appeared similar between the samples grown at 180 °C and 200 °C.
 |
| | Fig. 4 TEM images of samples grown at different reaction temperatures times in the presence of ascorbic acid. (A) 120 °C, (B) 140 °C, (C) 160 °C, (D) 180 °C, (E) 200 °C, (F) 2 h and (G) 10 h. The left-hand images have a magnification of 50000× and the right-hand images have a magnification of 200000×. | |
Fig. 4F and G show TEM images of samples obtained with different times (i.e., samples AA1.5/2 h and AA1.5/10 h). The reaction time had little effect on the morphology of the porous structures. With a reaction time of 2 h, the TEM images reveal a uniform spherical nanostructures, approximately 0.8 μm in diameter (see Fig. 4F), which is consistent with the FESEM results. The edges of the spheres appeared to be thick but relatively smooth. For the longer reaction time of 10 h, the uniformity of spheres remained almost constant, but the edges became thinner (see Fig. 4G). Therefore, the reaction time appeared to be significantly affecting porosity of the microspheres.
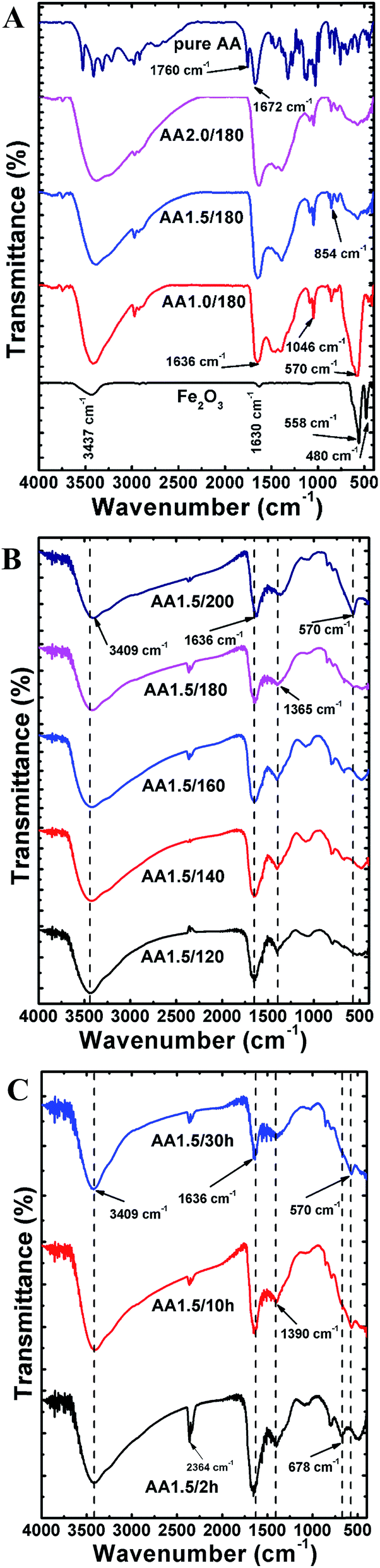
FTIR analysis
To analyze the chemical composition and interactions between molecules, we recorded FTIR spectra of the micro/nanospheres in range 400–4000 cm−1. Fig. 5A shows FTIR spectra of samples grown with various quantities of AA (i.e., samples AA0/180, AA1.0/180, AA1.5/180 and AA2.0/180). The FTIR spectrum of pure AA powder is also shown for comparison. The structure without AA exhibited two strong peaks at 480 and 558 cm−1, which are attributed to vibrations of Fe–O bonds in the hematite nanostructure.20 In addition, two broad bands were observed at 1630 and 3437 cm−1, which can be ascribed to water adsorbed on the surface of the hematite.36 With AA, the main characteristic vibrational peak of the magnetite phase was found at 570 cm−1, which indicates phase conversion from hematite to magnetite.37 As the quantity of AA increased, the intensity of the magnetite peak became weaker, which is indicative of the amorphous nature of iron oxide along with a reduced fraction of the magnetite phase. These results are in agreement with the XRD patterns, and the structures obtained with AA exhibited many common characteristic peaks form the spectrum of pure AA. In particular, the peaks at 1760 and 1672 cm−1 correspond to stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC, respectively.38 The strong absorption band at 1636 cm−1 under hydrothermal conditions is attributed to a CC stretching mode, which indicates carbonization of AA at high temperatures.39 It is interesting that the location of the CC bond was redshifted compared with the spectrum for pure AA, indicating a strong interaction between AA and the iron oxide particles formed during the hydrothermal process.40 Other common bands were found to in the range 854–1046 cm−1, which can be ascribed to symmetric and asymmetric stretching vibrations of C–O–C and C–OH bonds.41 Furthermore, four major bands at 1322, 1125, 1030 and 744 cm−1 were aroused from stretching vibrations from C–H, C–O–C, C–O–H and C–C groups existed in pristine AA powder.38,42 The broad peak in the range 3000–3600 cm−1 was ascribed to the existence of –OH groups. Fig. 5B shows FTIR spectra of powders obtained at different reaction temperatures (i.e., samples AA1.5/120, AA1.5/140, AA1.5/160, AA1.5/180 and AA1.5/200), where all other parameters were fixed. All spectra exhibit two common peaks at 3409 cm−1 and 1636 cm−1, which are attributed to the stretching vibration modes of –OH and CC groups, respectively.43 The peaks related to C–O–C and C–O–H groups were found to be absent because of hydrothermal treatment. Interestingly, presence of peaks at 1365 and 744 cm−1 after reaction process indicated the carbonization of AA molecules. A further peak at 570 cm−1, which is related to the magnetite phase, was absent for powders processed at lower temperatures, and present for the samples obtained at 180 °C and 200 °C. This indicates that a crystalline magnetite phase only formed at temperatures of 180 °C or higher. As shown in Fig. 5C, the samples obtained with different reaction times (i.e., AA1.5/2 h, AA1.5/10 and AA1.5/30 h) exhibited two common peaks at 3490 cm−1 and 1636 cm−1, which are attributed to the stretching vibration modes of –OH and CC groups, respectively.44 Remarkably, the magnetite peak at 570 cm−1 was detected for samples with reaction times of 10 and 30 h only. This indicates that a relatively long period of time was required for the formation of the magnetite phase, and this peak was absent from samples formed with a short time, indicating that 2 h was not sufficient to form the crystalline phase of magnetite. Two peaks at 1390 cm−1 and 678 cm−1 related to C–H and C–C groups, revealed the carbonization of AA after hydrothermal treatment.
O and CC, respectively.38 The strong absorption band at 1636 cm−1 under hydrothermal conditions is attributed to a CC stretching mode, which indicates carbonization of AA at high temperatures.39 It is interesting that the location of the CC bond was redshifted compared with the spectrum for pure AA, indicating a strong interaction between AA and the iron oxide particles formed during the hydrothermal process.40 Other common bands were found to in the range 854–1046 cm−1, which can be ascribed to symmetric and asymmetric stretching vibrations of C–O–C and C–OH bonds.41 Furthermore, four major bands at 1322, 1125, 1030 and 744 cm−1 were aroused from stretching vibrations from C–H, C–O–C, C–O–H and C–C groups existed in pristine AA powder.38,42 The broad peak in the range 3000–3600 cm−1 was ascribed to the existence of –OH groups. Fig. 5B shows FTIR spectra of powders obtained at different reaction temperatures (i.e., samples AA1.5/120, AA1.5/140, AA1.5/160, AA1.5/180 and AA1.5/200), where all other parameters were fixed. All spectra exhibit two common peaks at 3409 cm−1 and 1636 cm−1, which are attributed to the stretching vibration modes of –OH and CC groups, respectively.43 The peaks related to C–O–C and C–O–H groups were found to be absent because of hydrothermal treatment. Interestingly, presence of peaks at 1365 and 744 cm−1 after reaction process indicated the carbonization of AA molecules. A further peak at 570 cm−1, which is related to the magnetite phase, was absent for powders processed at lower temperatures, and present for the samples obtained at 180 °C and 200 °C. This indicates that a crystalline magnetite phase only formed at temperatures of 180 °C or higher. As shown in Fig. 5C, the samples obtained with different reaction times (i.e., AA1.5/2 h, AA1.5/10 and AA1.5/30 h) exhibited two common peaks at 3490 cm−1 and 1636 cm−1, which are attributed to the stretching vibration modes of –OH and CC groups, respectively.44 Remarkably, the magnetite peak at 570 cm−1 was detected for samples with reaction times of 10 and 30 h only. This indicates that a relatively long period of time was required for the formation of the magnetite phase, and this peak was absent from samples formed with a short time, indicating that 2 h was not sufficient to form the crystalline phase of magnetite. Two peaks at 1390 cm−1 and 678 cm−1 related to C–H and C–C groups, revealed the carbonization of AA after hydrothermal treatment.
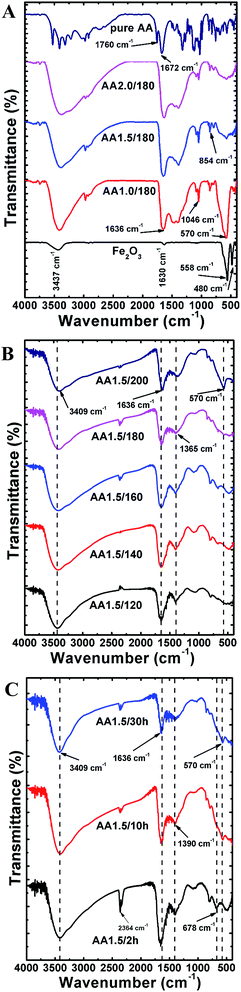 |
| | Fig. 5 (A) FTIR spectra of samples grown with various quantities of ascorbic acid. Spectra of ascorbic acid and a hematite sample obtained without ascorbic acid are also shown. (B) FTIR spectra of samples grown with various reaction temperatures. (C) FTIR spectra of samples grown with different reaction times. | |
XPS analysis
XPS measurements were carried out to investigate the oxidation states of Fe and the carbonization of AA under hydrothermal conditions (i.e., 180 °C, with a reaction of 5 h and 1.5 g of AA). The survey spectrum for the AA1.5/180 sample shown in Fig. S1A† reveals distinctive peaks at binding energies of 712, 531, 400 and 285 eV, which can be ascribed to Fe2p, O1s, N1s and C1s, respectively. The XPS spectrum of Fe2p shown in Fig. S1B† has two peaks centered at 724.8 and 710.8 eV, which are assigned to the presence of Fe2+(2p)1/2 and Fe2+(2p)3/2 levels, respectively.37 The deconvoluted Fe2+(2p)1/2 and Fe2+(2p)3/2 peaks could be assigned to Fe2+ (710.8 eV, 722.8 eV and 724.4 eV) and Fe3+ (709.2 eV, 712.6 eV, 725.9 eV) oxidation states of iron.19 The additional peaks at 727.5 eV and 715.9 eV were ascribed to carbonized phase of iron from iron carbonate, and this agrees with XRD data. The locations of these peaks indicate a similar binding energy to that reported previously,44 and which indicates the formation of a magnetite phase. Moreover, the absence of a charge-transfer satellite peak at ∼219 eV in the Fe2p spectrum indicates the magnetite phase of iron oxide. Fig. S1C† shows an XPS spectrum of the O1s peak centered at 531.2 eV, which is attributed to the O–Fe bond, and indicates the existence of a magnetite phase in the spherical nanostructures.45 Two additional peaks at around 529.4 and 532.9 eV could be aroused from surface oxygen species and hydroxyl groups, respectively.20 As shown in Fig. S1D,† the core level XPS spectrum of C1s exhibits six distinctive peaks, which are indicative of various chemical carbon states inside the porous structure. Among these, the two major peaks at 284.6 and 286.1 eV are assigned to C–C with sp2 hybridization, and to C–O with sp3 hybridization, respectively.46 The three lower intensity peaks at 287.6, 289.1 and 290.6 eV correspond to the existence of CO, O–CO and C–OH groups, respectively.47,48 Finally, the peak at 283.2 eV corresponds to a contribution from another source of carbon or interactions between Fe and C, which also indicates the presence of carbide particles.49 These XPS results therefore indicate the formation of a magnetite phase in the presence of AA, as well as carbonization, which resulted in the growth of carbon particles.
Magnetic properties
To investigate the magnetic properties of the nanostructures formed with different conditions, field-dependent magnetization curves were measured at room temperature in the range −10 to +10 kOe using a VSM. Fig. S2A–C† show hysteresis curves for various samples, and Table TS1† lists the coercivity Hc, remnant magnetization Mr and saturation magnetization Ms. The non-zero values of Hc and Mr (except for the sample obtained with a reaction time of 2 h), indicates the existence of a room-temperature magnetic moment in the nanostructures. The nanostructures grown with the optimum amount of AA, at high temperature, and with the longer reaction time (i.e., samples AA1.5/180, AA1.5/200, AA1.5/10 h and AA1.5/30 h) had relatively large values of Ms (>11 emu g−1), which is suitable for magnetic separation of catalyst particles. However, the amorphous structures had almost non-magnetic properties (see enlarged Fig. S2B†). Moreover, the small value of Hc at zero applied magnetic field indicates that these nanostructures may be magnetically separable from water, and then re-dispersed when the field is removed. These micro/nanospheres exhibit sufficient room-temperature magnetization for re-usability and hence have potential applications for the removal of contaminants from water.
Possible growth mechanism of the mesoporous spheres
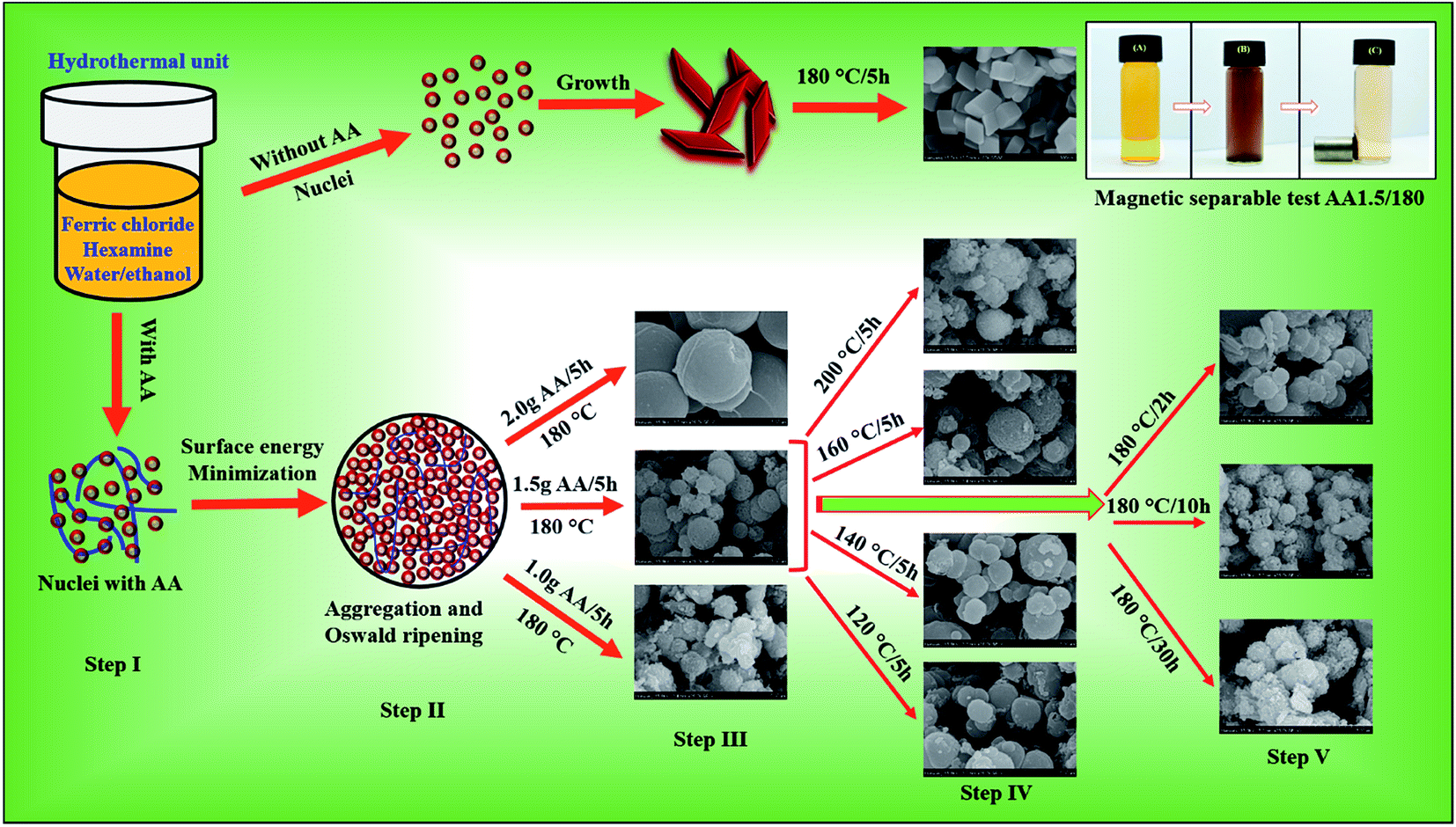
Differences in the growth rates along the different crystallographic axes determine the final shape and size of the nanostructures. Based on our experimental analysis, we propose the following formation mechanism for mesoporous spheres synthesized at temperatures below 200 °C via the hydrothermal method. The mesoporous structures were formed via Ostwald ripening under hydrothermal conditions. Initially, we carried out hydrothermal growth without AA, and observed that rhombohedral hematite structures were formed (Fig. 6, nucleation without AA). Clearly, therefore, AA is important in the formation of porous microspheres. There have been few reports of the synthesis of hematite, cobalt oxide or zinc oxide nanostructures using AA as a reducing or shape-directing agent.50–52 AA is known to be a reducing agent, and the rate of reduction is strongly dependent on the pH of the solution. In particular, in highly alkaline solutions, dissociation of AA is rapid, creating a large number of ascorbate anions and dianionic ascorbic ions.53 Moreover, Zhang et al. reported the synthesis of carbon nanoparticles using AA as a source of carbon via a hydrothermal method.39
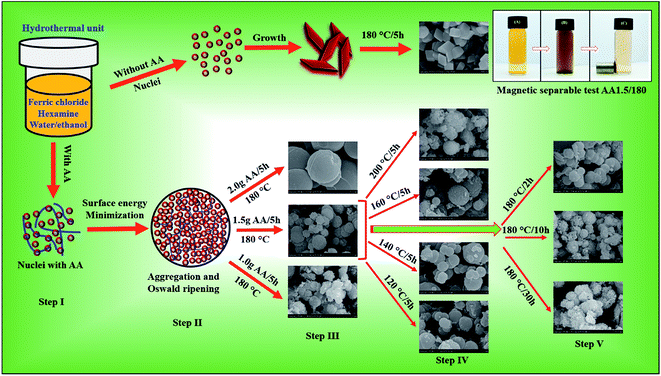 |
| | Fig. 6 Growth mechanisms of iron oxide nanostructure without and with AA, as well as photographs of the AO7 solution before and after magnetic separation of the mesoporous spheres. | |
Hydrothermal growth processes used to form inorganic nanostructure typically involve nucleation, minimization of the surface energy, aggregation and Ostwald ripening. Here, ferric chloride and hexamine were dissolved in a 50/50 vol% water/ethanol solvent at 180 °C for 5 h. In this process, hexamine is converted into HCHO and NH3 as follows:54
| | |
(CH2)6N4 + 6H2O → 6HCHO + 4NH3.
| (2) |
Following this reaction, the NH3 molecules react with water to generate hydroxyl ions (OH−), leading to an increase in the pH of the solution:
| | |
NH3 + H2O → NH4+ + OH−.
| (3) |
In addition to the above reactions, hydrolysis of iron chloride forms Fe3+ ions, which react with the hydroxyl groups, resulting in a hydroxide phase. The hydroxide phase is then converted into hematite as follows:55
| | |
Fe3+ + OH− → Fe(OH)3,
| (4) |
| | |
Fe(OH)3 → FeOOH + H2O
| (5) |
and
Via these reactions, in absence of ascorbic acid the final phase of iron oxide is hematite.
The effects of AA on the growth of iron oxide resulted in the growth of a magnetite phase with mesoporous spheres, rather then a hematite phase. Therefore, an investigation of the magnetite spheres is important to determine the role of AA during the hydrothermal process. As mentioned above, the presence of hexamine increased the pH of the solution, which led to dissociation of AA into ascorbate anion and dianionic ascorbic ions via the following reactions:
| | |
C6H8O6 → C6H7O6− + H+
| (7) |
and
| | |
C6H7O6− → C6H6O62− + H+.
| (8) |
In combination with this, in alkaline conditions, conversion of dianionic ascorbic ions with various intermediate steps occurs to form threonic acid (TA), which releases carbon dioxide (CO2) gas.53 The AA molecules reduce the Fe3+ ions to form Fe(OH)2 as follows:
| | |
2Fe3+ + C6H8O6 → 2Fe2+ + C6H6O6 + 2H+
| (9) |
and
| | |
Fe2+ + 2OH− → Fe(OH)2.
| (10) |
A phase transformation of the hydroxide iron oxide phases via reactions 5 and 10 leads to the formation of magnetite; i.e.,
| | |
FeOOH + Fe(OH)2 → Fe3O4 + 2H2O.
| (11) |
Several reactions occur during the growth of the magnetite spheres as a result of the presence of AA. To further investigate the nanopores in the spherical structures, we explored the effects of AA addition, the reaction temperature and the time period on the surface morphology.
As illustrated in Fig. 6 (nucleation with AA), structural evolution began with the nucleation and formation of Fe(OH)2 and Fe(OH)3 primary nanoparticles in the presence of AA (step I). This process was followed by aggregation and Ostwald ripening of small particles to reduce the surface free energy, resulting in the formation of spherical particles (step II). With an increase in pH, these hydroxide particles precipitated to form a goethite intermediate phase. During this aggregation and precipitation step, AA reduced the Fe3+ ions, decomposed into TA and released CO2, leading to the generation of nanopores inside the spherical structures. By varying the quantity of AA in the reaction solution, we could control the growth of the porous microspheres. The FESEM images of step III of the process indicate that the spheres grew uniformly with 1.5 g of AA. With 1 g of AA, polydispersed particles with a broader size distribution formed, indicating that 1 g of AA was not sufficient to form spherical structures. As we increased the quantity of AA to 2.0 g, most of the magnetite spheres disappeared, and new carbon spheres were formed. This larger quantity of AA led to the formation of carbon particles via carbonization. Hence we conclude that 1.5 g AA was optimal for the formation of nanoporous magnetite spheres.
The FESEM images of step IV of the process show magnetite spheres grown at different reaction temperatures. At low temperatures, only solid spheres formed, but at higher temperatures, the spheres were porous. Decomposition of hexamine and AA is slow at low temperatures; however, as the temperature increased, so did the rate of decomposition, leading to a more alkaline solution as well as rapid dissociation of AA. This leads to the formation of nanopores in the spherical structure, and hence to the growth of mesoporous spheres. At 200 °C, however, almost solid, compact spheres were observed because of the rapid crystal growth. We then varied the reaction time through 2, 10 and 30 h (step V), while maintaining the reaction temperature at 180 °C and the quantity of AA at 1.5 g. A short reaction time led to the formation of dense spheres, indicating that 2 h was insufficient to complete the reaction process. With 10 and 30 h, we found similar structures, but with a rougher surface than the sample with a reaction time of 5 h. We therefore conclude that 5 h is sufficient to complete the growth process and form porous microspheres. Additionally, we have proposed that the number of pores and surface roughness increased with temperature. Therefore, change in porosity was stemmed from rate of decomposition of AA with increasing temperature. Hence, temperature was crucial to grow porous microspheres with optimum porosity.
Specific surface area and pore size distribution
For effective removal of contaminants via adsorption processes, a large specific surface area is desirable, together with an optimal pore size distribution. We measured the BET surface area and porosity using N2 adsorption–desorption isotherms at 77 K for samples formed with different hydrothermal conditions. As shown in Fig. S3A–J,† all hysteresis curves could be categorized as type IV (as per the IUPAC nomenclature), which clearly indicates a mesoporous structure, with a large number of pores. Table TS2† lists details of the measured specific surface area for all of the mesoporous structures. The specific surface areas for spherical structures grown with AA (i.e., 18.27 m2 g−1 for sample AA1.0/180, 36.05 m2 g−1 for sample AA1.5/180 and 87.36 m2 g−1 for sample AA2.0/1800) were significantly larger than for the sample without AA (i.e., 5.54 m2 g−1 for sample A0/180). These results show that AA was crucial in increasing the specific surface area of the iron oxide samples. The mesoporous nature of isotherms could be ascribed to aggregation of secondary particles during the hydrothermal growth process. Furthermore, AA induced the formation of nanopores inside the spherical structures, leading to a high specific surface area. The specific surface areas for reaction temperatures of 120, 140, 160, 180 and 200 °C were 3.03 m2 g−1, 5.59 m2 g−1, 31.82 m2 g−1, 36.05 m2 g−1 and 30.03 m2 g−1, respectively. Clearly, the reaction temperature affected the specific surface area, and 180 °C was the optimum temperature. At low temperatures, particles formed with micron-scale sizes, and the absence of pores resulted in a low specific surface area. At high reaction temperatures, samples exhibited a large specific surface area because of the generation of a large number of nanopores.
Table TS2† lists the measured specific surface area of the magnetite samples grown with different reaction times. With a short reaction time of 2 h, the specific surface area was only 3.87 m2 g−1, which is smaller than that when AA was not used. This may be attributed to the random shape and micron-sized particles with the short reaction, as observed by TEM. With a longer reaction time of 10 h, the specific surface area increased to 33.07 m2 g−1, but decreased to 16.05 m2 g−1 after 30 h. With longer reaction times, the spherical structure became compacted due to the rapid grain growth, which reduced the number of pores existed in the microspheres. The insets of Fig. S3† show the pore size distribution measured using the BJH method. Most of the samples exhibited a wide pore size distribution, due to the large number of pathways for the effective transport of dye molecules towards the internal surfaces of the microspheres. This is expected to be useful for the effective adsorption of pollutants and removal of toxic compounds.
Removal performance of porous structures via adsorption process
Water pollution is a particularly important environmental concern because of its serious effects on human health. Furthermore, growing water scarcity requires high-performance processes to purify contaminated water. Conventionally, physical (adsorption and filtration) and chemical (precipitation, catalysis and oxidation) methods have been used to remove toxic elements and compounds from water.56,57 In particular, adsorption is a relatively straightforward, single-step and low-cost process for the removal of carcinogenic contaminants from water using porous nanomaterials. From the results described above, it is clear that our porous microspheres are suitable for removal of contaminants as part of wastewater treatment, as the large number of nanopores in the spherical structures provides channels to adsorb contaminants. Chromium(VI) and acid orange 7 are two carcinogenic pollutants emitted from cosmetic and textiles industries into water. Their removal via adsorption is therefore important, and low-cost methods to do so are particularly attractive.
Removal of AO7
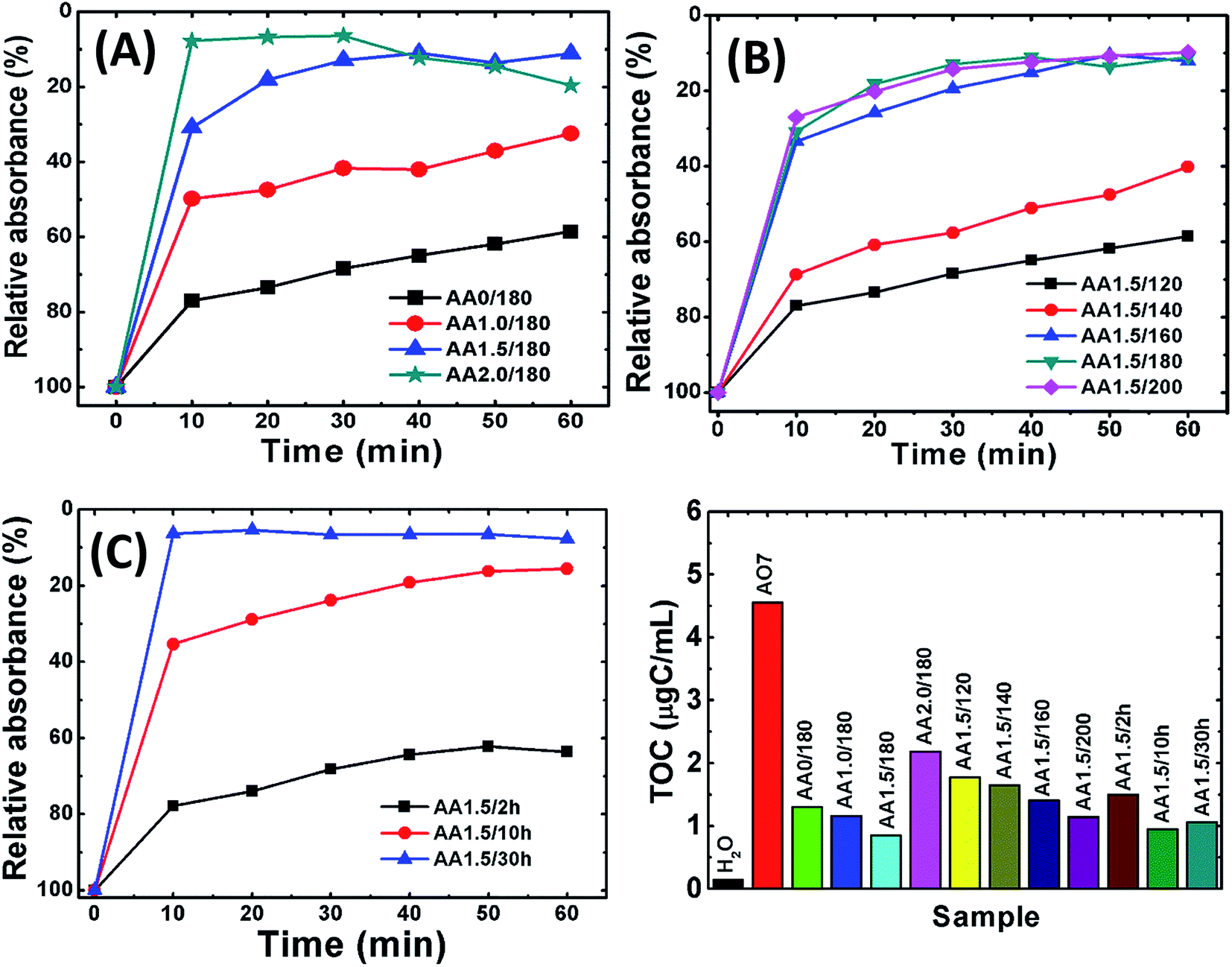
Initially, we measured the removal performance of the AA0/180, AA1.0/180, AA1.5/180 and AA2.0/180 powders obtained from growth solution without and with AA. Fig. 7A shows the relative absorbance of AO7 dye remained in the solution as a function of time. It is observed that almost 90% of dye was removed within 20 min for samples AA1.5/180 and AA2.0/180. This result clearly indicates that porous structures are useful to remove pollutants from water. Simultaneously, former structures have higher removal performance as compared with that of AA0/180 and AA1.0/180. Furthermore, sample AA2.0/180 exhibited similar performance to that of AA1.5/180, which may result from carbon spheres rather than the porous magnetite structure. Simultaneously, we conducted TG-DSC analysis of AA1.5/180 sample to find out the presence of carbon percentage inside magnetite structure. As shown in Fig. S4,† it can be seen that initial weight loss is attributed to evaporation of adsorbed water (from room temperature to 100 °C) and decomposition of carbon residue (from 100 to 500 °C). Afterwards, magnetite phase could be converted into hematite with complete oxidation and removal of carbon residue. The amount of carbon residue was calculated using following eqn (12):| |
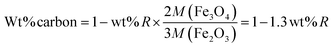 | (12) |
where wt% R is the remaining weight percentage after combustion, and M shows the molecular weight of the compound. The amount of carbon wt% from eqn (12) was found to be 58.38% for AA1.5/180 sample. Therefore, we could say that carbonization of AA resulted in the formation of carbon residue with magnetite structure, contributing to the removal of pollutant.
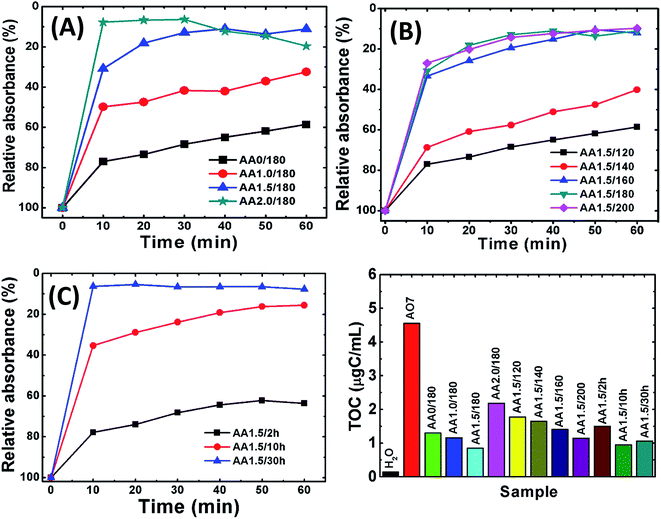 |
| | Fig. 7 The relative removal of AO7 using samples obtained with various reaction conditions. (A) AA0/180, AA1.0/180, AA1.5/180 and AA2.0/180; (B) AA1.5/120, AA1.5/140, AA1.5/160, AA1.5/180 and AA1.5/200; (C) AA1.5/2 h, AA1.5/10 h and AA1.5/30 h. (D) The TOC for all samples. The TOC of pure water and of AO7 are also shown for reference. | |
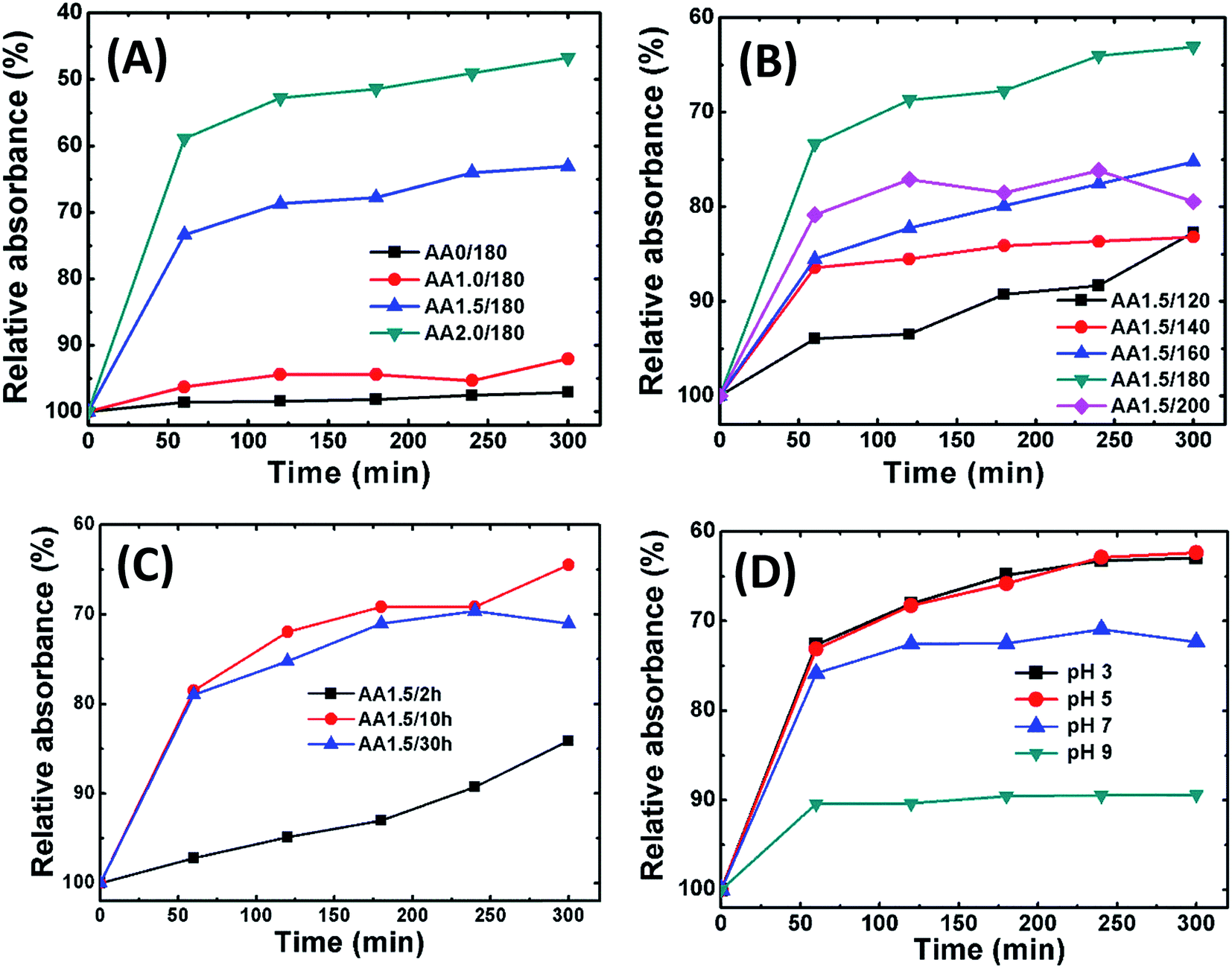
Fig. 7B shows the removal performance of magnetite powders obtained with AA at different reaction temperatures (i.e., samples AA1.5/120, AA1.5/140, AA1.5/160, AA1.5/180 and AA1.5/200). The different relative absorbance trends revealed that the reaction temperature is important in the growth of the porous structures. Interestingly, samples AA1.5/160, AA1.5/180 and AA1.5/200 exhibited better removal performance compared with samples AA1.5/120 and AA1.5/140. The superior performance for samples formed at high temperatures is attributed to the presence of a large number of nanopores, which can adsorb dye molecules. Fig. 7C shows the relative absorbance of AO7 solution as a function for samples AA1.5/2 h, AA1.5/10 h and AA1.5/30 h. We have found that the reaction time was also important, and that its removal performance improved with longer reaction times due to formation of relatively large number of pores. Longer reaction times induced more nanopores in the spherical structure, leading to effective removal of AO7 dye. Moreover, the present structures have shown better adsorption capacity than that of reported values shown in the literature.58–62 As shown in Table 1, nanoporous microsphere (A2.0/180 = 69.13 mg g−1) exhibited almost 16 times more adsorption capacity of AO7, compared to the rhombohedra structure (A0/180 = 4.61 mg g−1, Fig. S5A†). The obtained results confirm that present magnetic structure could be suitable for removal of AO7 from water with magnetically separable property.
Table 1 The removal performance of different materials reported in the literature and present work for AO7 dye
| Sr. no. |
Name of single component adsorbent |
Adsorption capacity (mg g−1) |
pH of solution |
Ref. no. |
| 1 |
Activated carbon coated ZnO nanoparticles |
32.13 |
3 |
58 |
| 2 |
Fe coated pumice |
27.56 |
9 |
59 |
| 3 |
Fe–Ni/CNTs |
25.60 |
4.5 |
60 |
| 4 |
Canola stalks |
25.1 |
2 |
61 |
| 5 |
Activated carbon |
44.0 |
8 |
62 |
| 6 |
Rhombohedral Fe2O3 (A0/180) |
4.61 |
7 |
Present work |
| 7 |
Nanoporous microspheres (A2.0/180) |
69.13 |
7 |
Present work |
Following the removal of AO7, we measured the TOC of the solution. Fig. 7D shows the TOC of AO7 solutions for iron oxide nanostructures obtained without and with AA. The TOC of distilled water and pure AO7 solution are also shown for reference. After 1 hour, for all samples the TOC decreased as compared with that of the AO7 solution. In particular, for sample AA1.5/180, the TOC was less than 0.8 μgC mL−1, which represents a decrease of ∼80% compared with the initial concentration of 4.5 μgC mL−1. Further, the removal capacity of porous structure independent of surface area was determined and summarized in Table TS2.† Clearly, it confirms that the porous structures show better performance compared to pristine structure. This good performance is attributed to the presence of a large number of nanopores, which result in a large specific surface area.
Removal of Cr(VI)
Fig. 8A shows adsorption isotherm curves of Cr(VI) on iron oxide nanostructures grown with and without AA (0, 1.0, 1.5 and 2.0 g) as a function of time. From these isotherms, it appears that the adsorption kinetics of Cr(VI) is a two-step process: the initial rate of removal of Cr(VI) was high, followed by a much slower subsequent removal rate, which eventually saturated. Interestingly, sample AA2.0/180 demonstrated the best performance; however, this sample also exhibited a relatively large number of carbon spheres, and hence the contribution of these may have been important. Because we focus on magnetite structures, we maintained the AA at 1.5 g or lower in further experiments. Fig. 8B shows the relative absorbance of Cr(VI) as a function of time for samples at different reaction temperatures (i.e., AA1.5/120, AA1.5/140, AA1.5/160 and AA1.5/200). Sample AA1.5/180 exhibited the best removal performance; therefore, 180 °C appears to be the optimum temperature to form spherical nanostructures with a large number of nanopores. Fig. 8C shows the relative absorbance as a function of time for samples formed using different reaction times. The reaction time is also significant in the removal of Cr(VI). The relative adsorption of Cr(VI) increased rapidly for samples AA1.5/10 h and AA1.5/30 h, and considerably more slowly for sample AA1.5/2 h. From these results, it appears that the adsorption of Cr(VI) was mainly determined by the nanopores in the magnetite structure. With a short reaction time, there were relatively few pores, leading to poor removal performance.
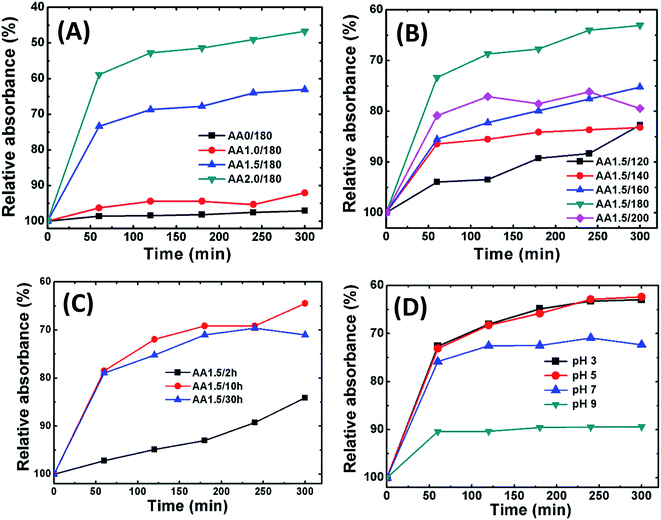 |
| | Fig. 8 The relative removal of Cr(VI) using samples obtained with various reaction conditions. (A) AA0/180, AA1.0/180, AA1.5/180 and AA2.0/180; (B) AA1.5/120, AA1.5/140, AA1.5/160, AA1.5/180 and AA1.5/200; (C) AA1.5/2 h, AA1.5/10 h and AA1.5/30 h. (D) Removal performance of AA1.5/180 sample in the pH range of 3 to 9. | |
Fig. 8D shows the relative absorbance of Cr(VI) removal of AA1.5/180 sample at different pH of solution (pH in the range 3–9). The adsorption was greater under acidic conditions. It has been reported that, under the acidic conditions, the surface of magnetite become positively charged, which improves the adsorption of the (negatively charged) Cr(VI) (CrO42−) species.63 Moreover, as the pH of solution increases, deprotonation of magnetite surfaces occurs, leading to a net negative charge and hence reduction in the rate of adsorption of Cr(VI).64 Moreover, the maximum adsorption capacity of A0/180 (10.67 mg g−1) and A2.0/180 (96.98 mg g−1) samples were calculated using Langmuir–Freundlich model, as summarized in Table 2 (Fig. S5B and C†). Clearly, it is seen that present nanostructures exhibited superior performance than that of previous work.65,66 These results show that Cr(VI) removal occurs via physical adsorption on the surface of the porous spheres.
Table 2 Comparative values of different nanostructures of iron oxide reported in the literature and present work for Cr(VI)
| Sr. no. |
Name of single component adsorbent |
Adsorption capacity (mg g−1) |
pH of solution |
Ref. no. |
| 1 |
Hollow urchin like FeOOH |
58.97 |
3 |
13 |
| 2 |
Porous Fe3O4 hollow microspheres/graphene oxide |
32.33 |
4.5 |
15 |
| 3 |
Flower-like Fe2O3 |
30 |
3 |
27 |
| 4 |
FeOOH coated Fe2O3 nanoparticles |
25.83 |
2.5 |
65 |
| 5 |
Mesoporous Fe2O3 |
15.6 |
2.5 |
66 |
| 6 |
Rhombohedral Fe2O3 (A0/180) |
10.67 |
5 |
Present work |
| 7 |
Nanoporous microspheres (A2.0/180) |
96.98 |
5 |
Present work |
Recycle stability
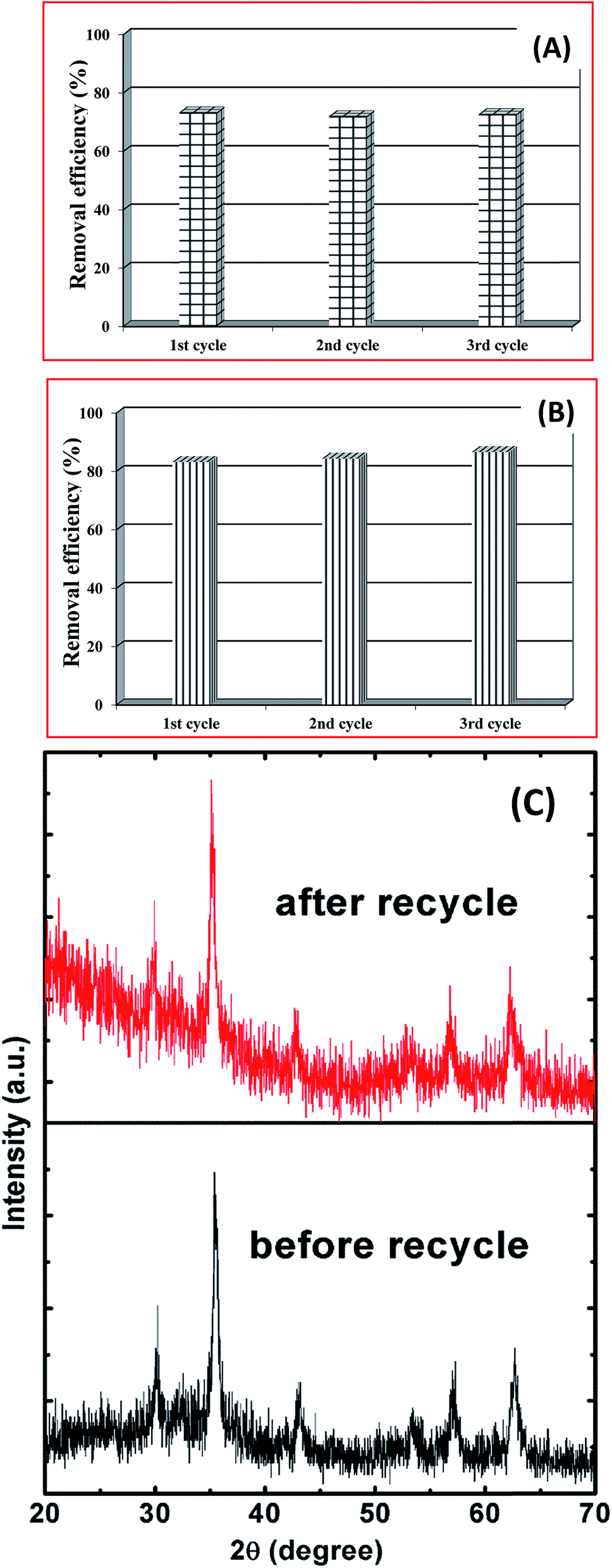
The recycle stability of the porous spheres is important for practical wastewater treatment applications. The main purpose of recovery and reuse of magnetite particles is to reduce the overall costs and energy consumption. The magnetic separability was investigated following the adsorption process using an externally applied magnetic field near the bottom of a glass vial (inset of Fig. 6). Initially, the color of the Cr(VI) solution was yellow, and following dispersion of the magnetite spheres it became black. When we applied the magnetic field, most of the dispersed particles were attracted and the solution became colorless. This shows that the spherical magnetite structures exhibit effective adsorption and magnetic separability.
The same powder was then recycled using the magnetic field, and re-dispersed for a second absorption cycle, following a removal (desorption) of the adsorbed Cr(VI) was using 0.10 M NaOH. This experimental procedure was repeated for three consecutive cycles, as shown in Fig. 9A. We observed no significant changes in the adsorption of Cr(VI), which indicates good stability and magnetic separability. Similar results were found for the case of removing AO7 dye up to three consecutive cycle as shown in Fig. 9B. Here, the adsorbed AO7 dye was removed by shaking and washing with distilled water several times. Following three cycles, the powder was separated and XRD patterns were measured, as shown in Fig. 9C. The XRD patterns following these cycle stability tests did not differ significantly from the original pattern for the AA1.5/180 sample. These results show that the magnetite porous spheres grown with AA have potential applications for the removal of Cr(VI), and are stable with reuse.
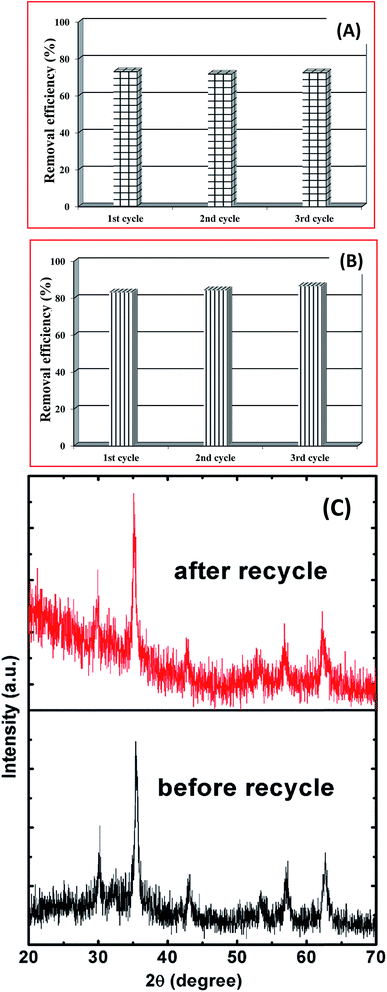 |
| | Fig. 9 The results of reusability test, (A) showing the removal efficiency of Cr(VI) for the 1st, 2nd and 3rd cycles of the magnetic separability tests, (B) showing the removal efficiency of AO7 for the 1st, 2nd and 3rd cycles, and (C) XRD patterns of sample AA1.5/180 before and after the cycling test. | |
Conclusion
We have described a facile, template-free approach for the growth of porous magnetite microspheres using a hydrothermal process. The presence of AA during the growth process was crucial for the formation of nanopores inside the spherical structures. We formed structures with different porosity and size distributions by systematically varying the growth conditions, and observed that the products obtained with 1.5 g AA at 180 °C and with a process duration of 5 h exhibited superior performance for the absorption of Cr(VI). The nanopores were important in the effective adsorption of pollutants, and the highly porous structures were found to provide efficient removal of contaminants from water. Magnetic separability was demonstrated, and cyclic absorption/separation/desorption tests revealed that the porous nanostructures were stable. We believe that these porous magnetite structures are suitable for practical applications in water treatment to remove wide range of contaminants.
Acknowledgements
This work was supported by the Energy Efficiency & Resources Core Technology Program of the Korea Institute of Energy Technology Evaluation and Planning (KETEP), granted financial resource from the Ministry of Trade, Industry & Energy, Republic of Korea. (No. 20142020103730), by the National Research Foundation of Korea (NRF) grant, funded by the Korean government (MEST) (2015R1A2A13027910) and the Human Resources Development program (No. 20154030200680) of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government Ministry of Trade, Industry and Energy.
References
- A. H. Smith, P. A. Lopipero, M. N. Bates and C. M. Steinmaus, Science, 2002, 296, 2145 CrossRef CAS PubMed.
- Y. Ao, J. Xu, X. Shen, D. Fua and C. Yuan, J. Hazard. Mater., 2008, 160, 295 CrossRef CAS PubMed.
- A. M. Balu, B. Baruwati, E. Serrano, J. Cot, J. G. Martinez, R. S. Varma and R. Luque, Green Chem., 2011, 13, 2750 RSC.
- S. Ye, L. Qiu, Y. Yuan, Y. Zhu, J. Xia and J. Zhu, J. Mater. Chem. A, 2013, 1, 3008 CAS.
- Y. Fu and X. Wang, Ind. Eng. Chem. Res., 2011, 50, 7210 CrossRef CAS.
- C. R. Oliveira and J. Rubio, Miner. Eng., 2006, 20, 552 CrossRef.
- L. Li, P. A. Quinlivan and D. Knappe, Carbon, 2002, 40, 2085 CrossRef CAS.
- G. Crini, Bioresour. Technol., 2006, 97, 1061 CrossRef CAS PubMed.
- J. S. Chen, C. Chen, J. Liu, R. Xu, S. Z. Qiao and X. W. Lou, Chem. Commun., 2011, 47, 2631 RSC.
- Y. Liu, L. Yu, Y. Hu, C. Guo, F. Zhang and X. W. Lou, Nanoscale, 2012, 4, 183 RSC.
- S. Xuan, W. Jiang, X. Gong, Y. Hu and Z. Chen, J. Phys. Chem. C, 2009, 113, 553 CAS.
- J. H. Pan, H. Dou, Z. Xiong, C. Xu, J. Ma and X. S. Zhao, J. Mater. Chem., 2010, 20, 4512 RSC.
- Y. Wang, J. Ma and K. Chen, Phys. Chem. Chem. Phys., 2013, 15, 19415 RSC.
- S. Xie, F. Lu, N. Sun, S. Liu, H. Jia and L. Zheng, CrystEngComm, 2014, 16, 9727 RSC.
- M. Liu, T. Wen, X. Wu, C. Chen, J. Hu, J. Lia and X. Wang, Dalton Trans., 2013, 42, 14710 RSC.
- W. Meng, W. Chen, L. Zhao, Y. Huang, M. Zhu, Y. Huang, Y. Fu, F. Geng, J. Yu, X. Chen and C. Zhi, Nano Energy, 2014, 8, 133 CrossRef CAS.
- X. Li, X. Huang, D. Liu, X. Wang, S. Song, L. Zhou and H. Zhang, J. Phys. Chem. C, 2011, 115, 21567 CAS.
- G. Tong, J. Guan and Q. Zhang, Mater. Chem. Phys., 2011, 127, 371 CrossRef CAS.
- K. Song, Y. Lee, M. R. Jo, K. M. Nam and Y. M. Kang, Nanotechnology, 2012, 23, 505401 CrossRef PubMed.
- X. Zhang, Y. Niu, X. Meng, Y. Li and J. Zhao, CrystEngComm, 2013, 15, 8166 RSC.
- C. Li, R. Younesi, Y. Cai, Y. Zhu, M. Ma and J. Zhu, Appl. Catal., B, 2014, 156–157, 314 CrossRef CAS.
- H. G. Yang and H. C. Zeng, J. Phys. Chem. B, 2004, 108, 3492 CrossRef CAS.
- T. J. Latempa, X. Feng, M. Paulose and C. A. Grimes, J. Phys. Chem. C, 2009, 113, 16293 CAS.
- Y. Wan, H. Yang and D. Zhao, Acc. Chem. Res., 2006, 39, 423 CrossRef CAS PubMed.
- G. Xi, C. Wang and X. Wang, Eur. J. Inorg. Chem., 2008, 3, 425 CrossRef.
- X. Liu, Y. Guo, Y. Wang, J. Ren, Y. Wang, Y. Guo, Y. Guo, G. Lu, Y. Wang and Z. Zhang, J. Mater. Sci., 2010, 45, 906 CrossRef CAS.
- C. Cao, J. Qu, W. Yan, J. Zhu, Z. Wu and W. Song, Langmuir, 2012, 28, 4573 CrossRef CAS PubMed.
- C. Wang, M. Cao, P. Wang, Y. Ao, J. Hou and J. Qian, Appl. Catal., A, 2014, 473, 83 CrossRef CAS.
- X. Li, B. Zhang, C. Ju, X. Han, Y. Du and P. Xu, J. Phys. Chem. C, 2011, 115, 12350 CAS.
- Q. Q. Xiong, J. P. Tu, Y. Lu, J. Chen, Y. X. Yu, Y. Q. Qiao, X. L. Wang and C. D. Gu, J. Phys. Chem. C, 2012, 116, 6495 CAS.
- F. Mou, J. Guan, H. Ma, L. Xu and W. Shi, ACS Appl. Mater. Interfaces, 2012, 4, 3987 CAS.
- W. Fan, W. Gao, C. Zhang, W. W. Tjiu, J. Pan and T. Liu, J. Mater. Chem., 2012, 22, 25108 RSC.
- Y. Wang, B. Zou, T. Gao, X. Wu, S. Lou and S. Zhou, J. Mater. Chem., 2012, 22, 9034 RSC.
- F. X. Ma, H. Hu, H. B. Wu, C. Xu, Z. Xu, L. Zhen and X. W. Lou, Adv. Mater., 2015, 27, 4097 CrossRef CAS PubMed.
- W. Omar, Chem. Eng. Technol., 2006, 29, 119 CrossRef CAS.
- Z. Liu, B. Li, B. Wang, Z. Yang, Q. Wang, T. Li, D. Qin, Y. Li, M. Wang and M. Yan, Dalton Trans., 2012, 41, 8723 RSC.
- W. Zhang, C. Kong and G. Lu, Chem. Commun., 2015, 51, 10158 RSC.
- J. Xiong, Y. Wang, Q. Xue and X. Wu, Green Chem., 2011, 13, 900 RSC.
- B. Zhang, C. Liu and Y. Liu, Eur. J. Inorg. Chem., 2008, 28, 4411 Search PubMed.
- Y. Yang, S. Han, G. Zhou, L. Zhang, X. Li, C. Zou and S. Huang, Nanoscale, 2013, 5, 11808 RSC.
- D. Maiti, U. Manju, S. Velaga and P. S. Devi, Cryst. Growth Des., 2013, 13, 3637 CAS.
- G. Xue, X. Huang, N. Zhao, F. Xiao and W. Wei, RSC Adv., 2015, 5, 13385 RSC.
- X. Li, X. Yu, J. He and Z. Xu, J. Phys. Chem. C, 2009, 113, 2837 CAS.
- F. Marquez, G. M. Herrera, T. Campo, M. Cotto, J. Duconge, J. M. Sanz, E. Elizalde, O. Perales and C. Morant, Nanoscale Res. Lett., 2012, 7, 210 CrossRef PubMed.
- Y. Wang, L. Zhang, X. Gao, L. Mao, Y. Hu and X. W. Lou, Small, 2014, 10, 2815 CrossRef CAS PubMed.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979 CrossRef CAS PubMed.
- X. Zhang, H. Tong, S. Liu, G. Yong and Y. Guan, J. Mater. Chem. A, 2013, 1, 7488 CAS.
- R. C. Pawar, Y. Pyo, S. H. Ahn and C. S. Lee, Appl. Catal., B, 2015, 176, 654 CrossRef.
- L. Zhu, L. Wang, N. Bing, C. Huang, L. Wang and G. Liao, ACS Appl. Mater. Interfaces, 2013, 5, 12478 CAS.
- W. Tan, Y. Yu, M. Wang, F. Liu and L. K. Koopal, Cryst. Growth Des., 2014, 14, 157 CAS.
- L. Zhao, K. Liao, M. Pynenburg, L. Wong, N. Heinig, J. P. Thomas and K. T. Leung, ACS Appl. Mater. Interfaces, 2013, 5, 2410 CAS.
- M. Raula, H. Rashid, T. K. Paira, E. Dinda and T. K. Mandal, Langmuir, 2010, 26, 8769 CrossRef CAS PubMed.
- Y. Lin and C. Liang, Environ. Sci. Technol., 2013, 47, 3299 CAS.
- R. C. Pawar, D. Choi and C. S. Lee, Int. J. Hydrogen Energy, 2015, 40, 767 CrossRef CAS.
- H. Singh, S. Bhagwat, S. Jouen, B. Lefez, A. A. Athawale, B. Hannoyer and S. Ogale, Phys. Chem. Chem. Phys., 2010, 12, 3246 RSC.
- H. Li, W. Li, Y. Zhang, T. Wang, B. Wang, W. Xu, L. Jiang, W. Song, C. Shu and C. Wang, J. Mater. Chem., 2011, 21, 7878 RSC.
- J. Hu, L. Zhong, W. Song and L. Wan, Adv. Mater., 2008, 20, 2977 CrossRef CAS.
- H. Nourmoradi, A. R. Ghiasvand and Z. Noorimotlagh, Desalin. Water Treat., 2014, 55, 252 CrossRef.
- B. Heibati, S. R. Couto, N. G. Turan, O. Ozgonenel, A. B. Albadarin, M. Asif, I. Tyagi, S. Agarwal and V. K. Gupta, J. Ind. Eng. Chem., 2015, 31, 124 CrossRef CAS.
- Y. Wang, S. Hsieh, C. Lee and J. Horng, Clean: Soil, Air, Water, 2013, 41, 574 CrossRef CAS.
- Y. Hamzeh, A. Ashori, E. Azadeh and A. Abdulkhani, Mater. Sci. Eng., C, 2012, 32, 1394 CrossRef CAS PubMed.
- S. Abera, N. Daneshvar, S. M. Soroureddin, A. Chabok and K. A. Zeynali, Desalination, 2007, 211, 87 CrossRef.
- J. Hu, G. Chen and I. M. C. Lo, Water Res., 2005, 39, 4528 CrossRef CAS PubMed.
- C. Mondal, M. Ganguly, J. Pal, A. Roy, J. Jana and T. Pal, Langmuir, 2014, 30, 4157 CrossRef CAS PubMed.
- J. Hu, I. Lo and G. Chen, Sep. Purif. Technol., 2007, 58, 76 CrossRef CAS.
- P. Wang and I. Lo, Water Res., 2009, 43, 3727 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02359b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.