DOI:
10.1039/C6RA02316A
(Paper)
RSC Adv., 2016,
6, 43080-43090
Substitution driven structural and magnetic transformation in Ca-doped BiFeO3 nanoparticles†
Received
26th January 2016
, Accepted 14th April 2016
First published on 25th April 2016
Abstract
Bi1−xCaxFeO3; (x = 0–0.20) nanoparticles were synthesized by tartaric acid based sol–gel route. X-ray diffraction and electron microscopy studies reveal the phase purity and nanocrystalline nature (45–90 nm) of Bi1−xCaxFeO3 samples. The Ca ion substitution driven structural transition from rhombohedral (space group R3c) to orthorhombic (Pnma) symmetry leads to enhancement in saturation magnetization due to the distorted cycloid spin structure (as also suggested by Mössbauer studies) and uncompensated surface spins which is accorded with electron paramagnetic resonance (EPR) studies as well. The ferromagnetic ordering contribution increases up to x = 0.15 samples with maximum saturation magnetization of 0.09 emu g−1 for x = 0.15 sample. The presence of high content orthorhombic phase for x = 0.20 sample results in the sharp decrease in the ferromagnetic component due to appearance of collinear antiferromagnetic ordering in agreement with EPR results. X-ray photoelectron spectroscopy confirmed the dominancy of Fe3+ oxidation states along with the shifting of the binding energy of Bi 4f orbital indicating the substitution of Ca2+ at Bi-site. Systematic change of Mössbauer parameters of nanoparticulate samples with Ca concentration are obtained by Mössbauer spectroscopy. The results of both one- and two-sextet fittings of the Mössbauer spectra provide evidence for destruction of the spin cycloid in Ca-doped BiFeO3 nanoparticles.
1. Introduction
Over the past few years, multiferroic materials, exhibiting simultaneous magnetic order and ferroelectric polarization in a single phase with strong coupling of electric, magnetic, and structural order parameters, have attracted much attention due to the emerging physics and potential applications in the special field of electromagnetic wave attenuation, data storage media, resistive switching, gas sensors and multi-state memories.1–7 However, naturally occurring single phase multiferroic materials are rare because of two reasons; (i) the ferroelectricity in ABO3 perovskite requires d0ness electronic configuration of transition metal ions, (ii) magnetism needs partially filled d orbitals of transition metal ions.2 “Multiferroic” properties can arise, however, when a polar cation such as Bi3+ or Pb2+ is present at the A-site of the perovskite and a magnetic cation is at the B-site. The off-center “lone pair” A-cation displacement may lead to polar superstructures with appreciable ferroelectricity, whereas ferro- or antiferromagnetic coupling of the transition metal cation spins leads to long-range magnetic order. BiFeO3 shows ferroelectricity (TC = 826–845 °C) and antiferromagnetism (TN = 370 °C) in single phase at room temperature and has rhombohedrally distorted perovskite structure with space group R3c, rhombohedral lattice parameters ar = 5.63 Å, αr = 59.35°, or alternatively, hexagonal parameters ahex = 5.58 Å, chex = 13.87 Å.7 Despite various interesting features, BiFeO3 exhibit various drawbacks, such as high leakage current, low magnetization, difficult to synthesize single phase. Hence for novel magnetoelectric applications ferroelectric and ferromagnetic properties must be enhanced. Many current investigations seek to suppress the spiral spin structure in BiFeO3 to enhance the magnetic properties and consequently to enhance the multiferroic properties.8,9 It is widely expected that the strategies based on decreasing the particle size (nanostructured materials) and divalent cations (Ba, Ca, Sr and Pb), transition metal ions (Co, Mn and Ni) and rare-earth metal ions, lanthanide series (Dy, Gd, Eu and Pr) substitution will have great impact on their magnetic, electric, optical and magnetoelectric properties.8–12 Recently, significant efforts have been made to understand the effect of chemical substitution of Ca on the crystal structure and magnetic properties of BiFeO3 ceramics. The ionic radius of Ca2+ ion (1.0 Å, CN 6) is quite smaller than that of Bi3+ ion (1.17 Å, CN 8), which can lead to large lattice distortion. The Ca2+ ion is magnetically inactive and the large distortion in crystal structure is expected to enhance the magnetization in BiFeO3 even for small doping concentration. These changes affect the degree of off-centering of the FeO6 octahedra and thus, also the multiferroic properties of BiFeO3. In polycrystalline samples synthesized by solid state route, Sardar et al. have shown that the effect of Ca2+ doping can be compared to that of hydrostatic pressure on BiFeO3, raising the Neel temperature TN.12,19 The substitution of Ca2+ ions at A site in BiFeO3 ceramics induced convergence between the magnetic Neel temperature TN and the ferroelectric Curie temperature TC and should lead to increased magneto-electric coupling. However, there are several contradictory reports on crystal structure and magnetic properties of Ca doped BiFeO3 ceramics with respect to the role of the Fe oxidation state (Fe2+, Fe3+ and Fe4+) or/and coordination and crystal structures. Charge compensation for the substitution of Ca2+ for Bi3+ in BiFeO3 ceramics could occur by the introduction of vacancies on oxygen sites or by oxidation of Fe3+ to Fe4+ giving theoretical solid solutions between BiFeO3 and CaFe3+O2.5 or CaFe4+O3, respectively. CaFeO3 can be produced under special conditions of high oxygen pressure and has the Pnma structure at room temperature. XANES and X-ray photoelectron spectroscopy results shows the wide range of Ca doped BiFeO3 samples prepared in air at 1 atm pressure have been found to contain only Fe3+. The clear implication is that the Ca substitution mechanism in BiFeO3 ceramics, charge compensation by oxygen vacancies. Subsolidus phase relations and strain data for the BiFeO3–CaFeO2.5 solid solution shows by Schiemer et al.13 There must be subtle structural/chemical influences which favor cycloidal, weakly ferromagnetic or purely antiferromagnetic ordering schemes, but this can be only a very minor variation. Second, ferroelectric ordering appears to be restricted to the rhombohedral structure. In BiFeO3 with low Ca-doping, there must be a balance between competing R- and M-point octahedral tilting, which would favor Pnma, and ferroelectric displacements coupling with R-point tilting, which favors R3c. This balance favors Pnma with addition of Ca ion. The evolution of crystal structure types with increasing the Ca content in BiFeO3 at room temperature is less clear. There are the lot of crystal structures reported with increasing the Ca content in BiFeO3 (rhombohedral, triclinic, cubic and orthorhombic) by different research groups.13–17 Schiemer et al. reported two phases at x = 0.1, one of which was metrically rhombohedral and the second metrically cubic, but also found evidence for other structure types with different superlattice repeats by electron diffraction.17 This two phase region directly contains the extrapolation of a linear fit to reported transition temperatures in the low Ca doping additional phases seems to relate to the onset of oxygen-vacancy ordering and that the variability reflects at least a degree of non-equilibrium from different sample preparation conditions and cooling rates. Recently, the most interesting Ca2+ doping experiments are of particular interest because they can be exploited to control the band-filling in insulating BiFeO3 thin films, by this means triggering an insulator–metal transition with varying composition.18,19 Simultaneously, in order to maintain the electrical neutrality of the system, acceptor dopant induced the oxygen vacancy, which is a kind of defect and can modify the electronic structure and the electrical conductivity. Furthermore, the increasing amount of oxygen vacancy would result in another interesting electrical conductive phenomenon-threshold switching (TS) effect.20 Among the various reasons of incompleteness on the studies of Bi1−xCaxFeO3 system includes the different values of x. The other reasons (like dependence of sample properties) strongly depends on method of preparation (i.e. solid state or sol–gel route) within the regimes of annealing, calcination and sintering of ceramic samples.12–19
Previous structural and Mössbauer studies were carried out mostly on bulk – Ca doped BiFeO3 samples to ravel structural transformation and its long-range ordering but few reports were available on Ca doped BiFeO3 nanoparticles. SQUID (or VSM) magnetic measurements provided information on net magnetization, but no further insight into the spin structure in nanoparticles using electron paramagnetic resonance studies were done. Our goal is to study Ca substitution dependent structural, vibrational and magnetic properties of BiFeO3 nanoparticles in details in order to determine the structural and magnetic transformations. For this purpose, nanoparticles were prepared by modified sol–gel route and systematically investigated for changes in the crystallographic structure, microstructure and magnetic properties. X-ray diffraction and Raman spectra confirmed the mixed two-phase structural state consisting of the rhombohedral and orthorhombic phases related to the distortions in a perovskite unit cell. X-ray Photo Spectroscopy (XPS) results confirmed the Fe3+ dominancy in Ca doped BiFeO3 nanoparticles. Prominence of weak ferromagnetism detected by VSM and EPR techniques was correlated with Ca substitution driven phase transformation originated from an incommensurate spin cycloidal modulated states to nearly collinear spin states. Mössbauer spectroscopy results showed the distorted cycloidal spin structure in Ca doped BiFeO3 nanoparticles.
2. Experimental details
2.1 Materials synthesis
Pure and Ca doped BiFeO3 nanoparticles were prepared by tartaric acid modified sol gel route. All the chemical (Sigma) reagents used as starting materials were analytic grade and do not require additional purification treatment. In a typical sol gel process for BiFeO3 nanoparticles, calculated amounts of bismuth nitrate (Bi(NO3)3·5H2O) and ferric nitrate (Fe(NO3)3·9H2O) were dissolved in deionized water. As Bi(NO3)3·5H2O undergoes decomposition into bismuth oxychloride in water, it was first taken in 50 ml deionized water and kept on stirring. During stirring dilute nitric acid was added drop by drop until a clear and transparent solution was obtained which confirmed the complete dissolution of Bi(NO3)3·5H2O then ferric nitrate solution was added. The stoichiometric amount of tartaric acid (C6H6O7) was further added for the complete combustion of the nitrates. In present report tartaric acid was used as chelating agent for the synthesis of Ca doped BiFeO3 nanoparticles. It is difficult to get phase pure BiFeO3 samples and several secondary phases (Bi2O3, Bi2Fe4O9 and Bi46Fe2O72) exist in the samples synthesized with citric acid as chelating agent. S. Gosh et al. and M. Arora et al. reported pure phase formation of BiFeO3 nanoparticles by using tartaric acid as chelating agent.21 The uniqueness of the tartaric acid as a chelating agent in synthesis of BiFeO3 nanoparticles probably resides in the formation of hetero-metallic polynuclear complexes in the solution, where reacting metal atoms come in close proximity. This occurs because of the presence of two carboxylate and two hydroxyl groups with proper orientation to form a polynuclear complex, which breaks on heating in the presence of concentrated HNO3 leading to the formation of pure phase BiFeO3 samples. The yellow transparent solution was stirred vigorously for 12 h over a hot plate kept at 60 °C. Further the transparent solution was dried for two days in an oven maintained at 120 °C to get the fluffy gel. Finally, the fluffy gel was sintered at different temperatures 400, 500 and 600 °C for two hours in an air ambience. Ca doped BiFeO3 samples with different Ca doping concentrations were prepared by adding the calculated amount of Ca(NO3)2·5H2O in the precursor solution keeping all other experimental conditions constant.
2.2 Characterization
The prepared samples were structurally characterized by X-ray diffractometer (Shimadzu 6000 XRD). The measurements were carried out at room temperature using Cu Kα radiation source (λ = 1.5406 Å, operated at 40 kV and 30 mA) and the data was collected in the range 2θ = 10° to 95° with the step size 0.02° and 1.2 seconds count time at each step. Fullprof (Version Feb. 2012) was used for Rietveld refinement of XRD data. Scanning electron microscopic images were recorded using a Zeiss Ultra Plus field-emission scanning electron microscope (FESEM) at 10 kV operating voltage. The TECNAI with LaB6 electron source was used for High Resolution Transmission Electron Microscope (HRTEM) operated at 200 kV to image pure and Ca doped BiFeO3 nanoparticles. The samples for the TEM analysis were prepared by placing a drop of dilute suspension on carbon coated copper grid. Raman spectroscopy was carried out in the backscattering configuration (LabRAM HR) with charge coupled device detector and with 325, 514.5 and 785 nm laser excitation sources. The laser power was kept below 2 mW in order to avoid any sample heating. To analyze the oxidation states of the constituents, X-ray photoelectron spectroscopy (XPS) was used with AXIS ULTRA spectrometer with Al (Kα) source. The magnetic measurements of samples at room temperature were carried out using vibrating sample magnetometer (Lakeshore VSM 7410). The magnetic resonance spectra were recorded with an EPR spectrometer (JES FA200 CW ESR Spectrometer) using X-band gun diode. The samples were rolled into cylindrical shapes by wrapping them in Teflon tapes. These samples were stuffed into a quartz capillary tube. The sample was placed at the centre of the resonant cavity placed between pole caps of an electromagnet. The magnetic field was scanned from 0–800 mT, while the resonance frequency (∼9.46 GHz) of the sample cavity was locked. Mössbauer effect measurements were carried out using a standard PC-based spectrometer equipped with a Weissel velocity drive operating in the constant acceleration mode.
3. Result and discussion
3.1 X-ray diffraction analysis
Fig. 1(a) illustrates the X-ray diffraction patterns of the BiFeO3 samples sintered at 400, 500 and 600 °C, respectively. It has been clearly observed that there is no BiFeO3 diffraction peaks except amorphous phase (2θ ∼ 30°) in the sample sintered at 400 °C. With increasing the sintering temperature to 500 °C, the XRD pattern expectedly shows the typical peaks of rhombohedral structure (JCPDS card number 71-2494) indicating the presence of crystalline BiFeO3 ceramics.22 With further increasing the sintering temperature to 600 °C, the intensities of BiFeO3 diffraction peaks are found to increase, suggesting improved nature of crystallization. Fig. 1(b) shows the X-ray diffraction patterns and its magnified view (near 2θ ∼ 31 and 40°) of Bi1−xCaxFeO3 (x = 0–0.20) samples sintered at temperature 600 °C. Diffraction peaks (104) and (110) are clearly separated for pure BiFeO3 sample, however, on increasing Ca content from x = 0.05 to 0.20, doublet (104) and (110) merged to a single peak and shifted toward higher 2θ values. Also, (113) and (006) peaks are suppressed with increasing Ca content. These results indicate that Ca doping in BiFeO3 samples results in compressive lattice distortion. With smaller Ca2+ ion occupying the A site, the Goldschmidt tolerance factor ((〈rA〉 + rO)/(√2(〈rB〉 + rO))), where 〈rA〉 and 〈rB〉 are the average ionic radii of A and B site cations, respectively; and rO is the ionic radius of oxygen will systematically decrease. The smaller the tolerance factor, the more severe is the buckling of the oxygen octahedra. This is due to the fact that the smaller A site ion cannot fill the empty space completely and instead the tilting and shrinking of the octahedra take place. The lattice distortion due to the octahedral tilt eventually suppresses the rhombohedral phase. This ensuing lattice distortion leads to evolution of lower symmetric orthorhombic phase. For detailed structural analysis, the Rietveld refinement of pure BiFeO3 nanoparticles is carried out by considering the rhombohedrally distorted perovskite structure with R3c space group while Ca doped BiFeO3 samples are successfully refined by assuming a mixture of the rhombohedral R3c and orthorhombic Pnma space groups. The Rietveld refined XRD patterns are shown in Fig. 1(c)–(f). The Bragg peaks are modeled with Thompson–Cox–Hastings pseudo-Voigt function and the background is estimated by linear interpolation between selected background points. The refinement of the structural parameters is continued till convergence is reached with goodness of fit (GoF) around 1.0.23 The various parameters for both phases obtained from refinement of XRD patterns are listed in ESI Table S1.† Lattice parameters and unit cell volume of R3c phase as well as Pnma phase decrease with increasing the Ca content in BiFeO3 samples due to smaller ionic radius of doping cation Ca2+ (1.0 Å) than that of host cation Bi3+ (1.17 Å). For the Pnma space group, when a and c lattice parameters shrink due to smaller size of doping cations, the lattice parameter b is under stress and hence it increases. The crystal structure of BiFeO3 (R3c) and Bi0.8Ca0.2FeO3 (Pnma) generated using the refined ionic positions obtained from Rietveld refinement from FullPROF are shown in Fig. 1(g) and (h). The contribution of the orthorhombic phase increases from 4.52% for x = 0.05 sample to 84.20% for x = 0.20 sample.
 |
| | Fig. 1 (a) XRD patterns of BiFeO3 precursor gel sintered at different temperatures. (b) XRD patterns of Bi1−xCaxFeO3 nanoparticles sintered at 600 °C. Rietveld refined XRD patterns of Bi1−xCaxFeO3 nanoparticles, (c) x = 0.0, (d) x = 0.10, (e) x = 0.15 and (f) x = 0.20. Schematic representation of expected crystal structures for Bi1−xCaxFeO3, (g) for x = 0.0, rhombohedral perovskite R3c structure and (h) orthorhombic Pnma structure (representation is based on the refined ionic positions obtained from Rietveld refinement). | |
3.2 FESEM and TEM analysis
The morphological analyses of the synthesized samples were done by the field emission scanning electron microscopy (FESEM) technique. FESEM micrographs of the fractured surface (as shown in Fig. 2) indicated that the synthesized pristine BiFeO3 and Bi1−xCaxFeO3 (x = 0.10, 0.15 and 0.20) samples have dense morphology of spherical grain size ranging between 45 and 90 nm.
 |
| | Fig. 2 FESEM micrographs of Bi1−xCaxFeO3 nanoparticles, (a) x = 0.0, (b) x = 0.10, (c) x = 0.15 and (d) x = 0.20. | |
Shape, size, lattice interplanar spacing and crystallinity of the synthesized nanostructured samples were further characterized using HRTEM technique. The TEM images of Bi1−xCaxFeO3 samples with x = 0.0, 0.10 and 0.15 are shown in Fig. 3(a)–(c), respectively. It is evident that spherical nanoparticles of size around 45–90 nm are observed for x = 0.0, 0.10 and 0.15 samples. The TEM images also indicate that nanoparticles size decreases with increasing Ca concentration in BiFeO3, indicating the initiation of lattice strain due to ionic size mismatch between Ca and Bi, further enhancing the rate of nucleation and reducing the particle growth. The selected area electron diffraction (SAED) pattern of Bi0.85Ca0.15FeO3 nanoparticles is also shown in Fig. 3(d). The diffraction pattern comprising of discrete rings indicates the polycrystalline nature of the sample. The simulated rings pattern of Bi0.85Ca0.15FeO3 nanoparticles with the diffraction intensity of peaks is also shown in Fig. 3(d). Fig. 3(e) clearly shows the lattice interplanar spacing of Bi0.85Ca0.15FeO3 nanoparticles, which indicates that fine nanoparticles are well crystallized into single crystals. The Fast Fourier Transform (FFT) using Gatan Microscopy Suite software has been employed to measure the interplanar spacing and the FFT image is shown in the inset of Fig. 3(e). Fig. 3(f) shows the lattice plane intensity profile corresponding to Fig. 3(e), confirming the interplanar spacing of 0.396 nm corresponding to (012) plane (R3c space group) of the Bi0.85Ca0.15FeO3 nanoparticles.
 |
| | Fig. 3 TEM images of Bi1−xCaxFeO3 nanoparticles (a) x = 0.0, (b) x = 0.10, (c) x = 0.15, (d) selected area diffraction pattern for x = 0.15 nanoparticles with simulated ring pattern, (e) high resolution TEM image for x = 0.15 nanoparticles; the inset represents the fast Fourier transform (FFT) of the image and (f) the FFT was used to measure the interplanar distance corresponding to (012) plane. | |
3.3 Raman analysis
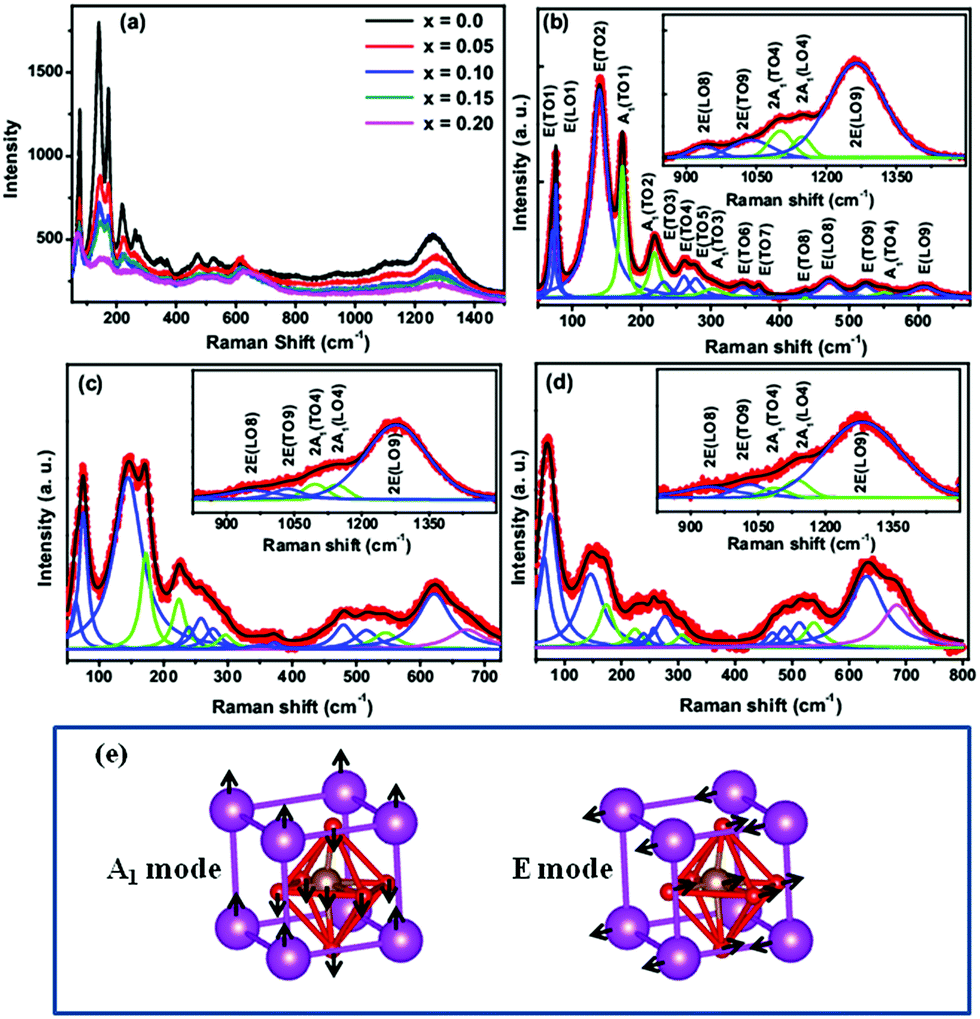
Fig. 4(a) shows the room temperature Raman spectra in the range 50–1500 cm−1 for Bi1−xCaxFeO3 (x = 0–0.20) nanoparticles. The assignment of observed Raman modes of distorted rhombohedral perovskite structure of BiFeO3 with space group R3c is based on first principal calculations carried out by Hermet et al.24 and experimentally reported FTIR data by Chen et al.25 and Raman data by Porporati et al. and Bielecki et al.26,27 The group theory analysis of the lattice vibrations in the R3c structure with two formula units per unit cell yields the following optic phonons:ΓR3c = 4A1(z,x2 + y2,z2) + 5A2(−) + 9E(x,y,x2 − y2,xy,xz,yz)
 |
| | Fig. 4 (a) Raman spectra of Bi1−xCaxFeO3 nanoparticles at room temperature. (b) Deconvoluted first order Raman modes for x = 0.0. Inset shows the second order modes for x = 0.0. (c) Deconvoluted first order Raman modes for x = 0.15. Inset shows the second order modes for x = 0.15. (d) Deconvoluted first order Raman modes for x = 0.20. Inset shows the second order modes of for x = 0.20. (e) Illustrations show the A1 and E vibration normal modes. | |
There are 13 [Longitudinal Optical (LO) or Transverse Optical (TO)] infrared and Raman active modes for R3c space group while the modes 5A2 are silent in the above irreducible representation equation. The Raman spectra of Bi1−xCaxFeO3 (x = 0–0.20) nanoparticles are fitted with multiple Lorentzian oscillators of the form 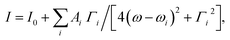 where i is the peak number, I0 accounts for the background intensity, ωi is the center frequency, Γi is the full width at half maxima (FWHM), and Ai is the area of ith peak. The fitted spectra for Bi1−xCaxFeO3 (x = 0.0, 0.15 and 0.20) nanoparticles are shown in Fig. 4(b)–(d), respectively. The observed modes position and FWHM of E and A modes for Bi1−xCaxFeO3 (x = 0–0.20) nanoparticles are summarized in ESI Table S2.† First Raman active mode near 75 cm−1 splits into two modes E(TO1) and E(LO1). Similarly both LO and TO modes are present for E(8) and E(9). The Raman modes doublet E(TO1), E(LO1) near 75 cm−1, E(TO2) mode at 139.2 cm−1 and A1(TO1) mode at 172.3 cm−1 are due to the displacement of Bi atoms. This displacement is caused by the activation of lone pair 6s2 electrons of Bi3+ along the c-axis of hexagonal unit cell. The Raman modes E(TO1)/E(LO1) provide the key information about the magnetoelectric coupling in BiFeO3 due to its strong polar character and further regulates the dielectric constant of the material. The shifting of the first order modes E(LO1)/E(TO1) with increasing the Ca content in BiFeO3 samples is associated with strain via phonon deformation potentials α, β and γ,
where i is the peak number, I0 accounts for the background intensity, ωi is the center frequency, Γi is the full width at half maxima (FWHM), and Ai is the area of ith peak. The fitted spectra for Bi1−xCaxFeO3 (x = 0.0, 0.15 and 0.20) nanoparticles are shown in Fig. 4(b)–(d), respectively. The observed modes position and FWHM of E and A modes for Bi1−xCaxFeO3 (x = 0–0.20) nanoparticles are summarized in ESI Table S2.† First Raman active mode near 75 cm−1 splits into two modes E(TO1) and E(LO1). Similarly both LO and TO modes are present for E(8) and E(9). The Raman modes doublet E(TO1), E(LO1) near 75 cm−1, E(TO2) mode at 139.2 cm−1 and A1(TO1) mode at 172.3 cm−1 are due to the displacement of Bi atoms. This displacement is caused by the activation of lone pair 6s2 electrons of Bi3+ along the c-axis of hexagonal unit cell. The Raman modes E(TO1)/E(LO1) provide the key information about the magnetoelectric coupling in BiFeO3 due to its strong polar character and further regulates the dielectric constant of the material. The shifting of the first order modes E(LO1)/E(TO1) with increasing the Ca content in BiFeO3 samples is associated with strain via phonon deformation potentials α, β and γ,
| ΔωE = α(εxx + εyy) + β(εzz) + γ(εxx − εyy) |
where
εxx,
εyy are the normal strains in the plane and
εzz in the
c-axis direction, respectively.
28–30 The suppression and shifting of Raman mode (ETO
2) related to Bi–O bond towards the higher wavenumber with increasing Ca content in BiFeO
3 is an indication of structural phase transition from rhombohedral to orthorhombic.
27 The other interesting feature in the Raman spectra is the relative increase in the intensity of A
1(TO
1) mode in comparison with the E(TO
2) with increasing the Ca content in BiFeO
3 samples, which is an evidence for the spin dependent scattering mechanism with the magnetic anisotropy.
29 In the case of magnetically ordered system, the intensity of the Raman mode can be given by the relation,
where
R is the spin independent part,
M is the magnetic moment, 〈
SiSj〉/
S2 is the nearest neighbor spin correlation function.
28,29 The enhancement of A
1(TO
1) intensity in comparison with the E(TO
2) with increasing the Ca content in BiFeO
3 is due to different contributions from the correlation function 〈
SiSj〉/
S2 in different directions, resulting from the change in magnetic ordering owning to the spin reorientation transitions with increasing the Ca content in BiFeO
3 samples. The next mode with A
1(TO
2) symmetry at 218.4 cm
−1 is soft oxygen mode. The A
1(TO
1) and A
1(TO
2) modes are due to Bi–O and Fe–O bonds. The modes A
1(TO
1), A
1(TO
2), E(TO
3), E(TO
4) and higher wave number modes are related to the Fe–O bond. The mode E(TO
5) at 278.1 cm
−1 is caused by atomic vibrations in the oxygen planes. The higher wave number modes above 250 cm
−1 are related to the oxygen motion.
Fig. 4(e) illustrates the A
1 and E vibration modes. The change in the position and FWHM of the modes with increasing Ca content in BiFeO
3 indicate the lattice distortion and structural phase transition.
The second-order Raman phonon modes of Bi1−xCaxFeO3 (x = 0.0, 0.15, 0.20) nanoparticles are shown in the inset of Fig. 4(b)–(d), respectively. There are five vibrational modes from 900 cm−1 to 1400 cm−1. These second-order modes labeled as 2E(LO8), 2E(TO9), 2A1(TO4), 2A1(LO4) and 2E(LO9) are the combination modes and overtones produced by the first-order modes positioned between 450 and 630 cm−1.27,31,32 These second order phonon modes are assigned to long and short Fe–O (defined as Fe–O1 and Fe–O2) bonding, respectively, where O1 are axial and O2 are equatorial ions.32 The Fe–O2 is related to the octahedral rotation critical to weak ferromagnetic behavior in BiFeO3. The structural distortion due to Ca doping should influenced Raman modes due to the spin phonon coupling in these samples. The intensity of second order phonon modes of Ca doped BiFeO3 samples decreases as compared to pure BiFeO3 sample indicating the change in rotation of oxygen octahedral critical to weak ferromagnetism through superexchange interaction.32 The variation of intensity of these second overtones predicts that magnetization might enhance with Ca content in BiFeO3, indicating the spin-two phonon coupling in these samples. The second order phonon peaks are associated with the strong spin lattice coupling arising from the interaction between the adjacent magnetic sublattices.
Fig. 5(a)–(c) shows the Raman scattering spectra of Bi1−xCaxFeO3 (x = 0.05, 0.15 and 0.20) nanoparticles measured by using three laser sources of different energy values (3.25 eV, 2.41 eV and 1.58 eV). The noisy spectra were observed due to the direct electronic transitions from valance band to conduction band when the laser source of 325 nm (3.81 eV) is used, which is ∼1.6 eV higher than the band gap value (2.2 eV) of the BiFeO3 nanoparticles.14,37 The intensity of the second order phonon modes at 1255 cm−1 is of the same order as that of the corresponding fundamental modes when laser source of 514.5 nm (2.41 eV) is used which is around 0.2 eV higher than the band gap value. The intensity of the second order modes around 1255 cm−1, measured by using the laser source of 7 85 nm (1.58 eV), is negligible because it does not have enough energy to promote direct and indirect electronic transitions. Another remarkable finding is a notable intensity variation of the fundamental modes ∼218 cm−1 measured by using 325 nm, 514.5 nm and 785 nm laser sources. It is important to mention that the intensity variations observed in some vibrational modes are exclusively related to the atoms taking part in the electronic transition in the resonance Raman phenomenon.37 The Raman mode ∼218 cm−1 is related to the Fe (3d) atoms. These facts could mean that when the energy of the laser source is sufficient to promote electronic transition from the valence band to the lower conduction band in BiFeO3, a perturbation occurs in the electronic density of the 3d orbitals of the Fe atoms.37 It consequently changes the polarizability of iron atoms resulting in enhancement of the intensity of 218 cm−1 mode. These results confirm that electronic transitions take place under excitation with 325 and 514.4 nm laser radiations, allowing the resonance effect.
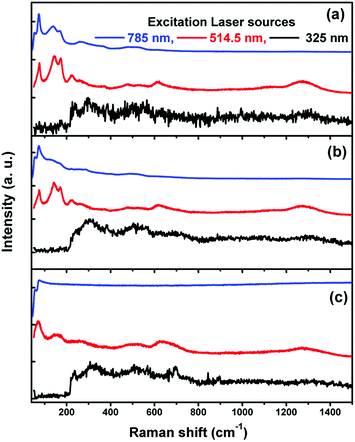 |
| | Fig. 5 Raman spectra of Bi1−xCaxFeO3 sample employing three different lasers as excitation sources at room temperature, (a) x = 0.05, (b) x = 0.15 and (c) x = 0.20. | |
3.4 X-ray photo spectroscopy (XPS) analysis
In order to identify the elements, chemical shift, oxidation state of elements and oxygen vacancies of the Ca doped BiFeO3 samples, detailed XPS analysis has been carried out. The narrow scan XPS spectra of Bi 4f, Fe 2p, Ca 3s and O 1s lines for Bi1−xCaxFeO3 (x = 0.10 and 0.15) samples are shown in Fig. 6. Fig. 6(a) shows that Bi 4f doublet consisting of two peaks at 158.8 and 164.1 eV, which are from Bi–O bonds. The moderate shifting of Bi 4f peaks towards the higher binding energy with increasing x indicates the substitution of Ca2+ ions at Bi3+ sites in BiFeO3 lattice. The chemical shift in Bi 4f7/2 and Bi 4f5/2 peaks arises due to variation in electro-negativity of Bi, Ca, Fe and O elements. The covalency/ionicity of Bi–O, Ca–O and Fe–O bonds have been calculated for x = 0.10 and 0.15 samples. The fraction of covalency (Fc) is estimated from the difference in anion and cation electronegativity values (ΔEN) as: Fc = exp(−(ΔEN)2/4), while the fraction of ionicity is calculated by: Fi = (1 − Fc).33,34 According to the electronegativity values of Bi, Ca, Fe and O elements, the Fc and Fi values for Bi–O, Ca–O and Fe–O bonds are calculated. The fraction of ionicity (Fi) for Ca–O bond (0.77) is much larger than that for Bi–O bond (0.40). This indicates that the bonding energy of (Bi/Ca–O) bond in the oxygen octahedron would be larger than that of pure Bi–O bond which results in slight shifting of 4f peaks towards the higher binding energy side with increasing Ca content in BiFeO3 samples. Fig. 6(b) shows Fe 2p XPS core spectra of Bi1−xCaxFeO3 samples with x = 0.10 and 0.15. The Fe 2p doublet consists of two wide peaks of Fe 2p3/2 ∼ 710.1 eV and Fe 2p1/2 ∼ 723.5 eV for x = 0.10, which are mainly ascribed to Fe–O bonds. Spin–orbit splitting energy is equal to 13.4 eV, which is comparable to the theoretical value of Fe 2p (13.6 eV) for Fe2O3. The peak Fe 2p1/2 for Fe3+ oxidation state is approximately 13.6 eV as discerned from the binding energy of Fe 2p3/2 appear at 710.7 and 709.98 eV for x = 0.10, 0.15 samples, respectively.34 In general, satellite peak appears at 8 eV above 2p3/2 for Fe3+. In both samples, a satellite peak is observed at ∼7.5 eV above Fe 2p3/2, which confirms the dominant 3+ oxidation state of Fe. According to the fitting, the compositional ratio of Fe2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe3+ is calculated as 33.3:66.7 and 39.9:60.1 for x = 0.10, 0.15 respectively. The Ca 2p spectra deconvoluted into two wide peaks of Ca 2p3/2 ∼ 346.26 eV and Ca 2p1/2 ∼ 349.81 eV for x = 0.10, 0.15 are shown in Fig. 6(c), which are mainly ascribed to Ca–O bonds.35 Fig. 6(d) shows the O 1s peak is de-convoluted into three peaks; the lower binding (LB) energy, medium binding (MB) energy and higher binding (HB) energy for x = 0.10, 0.15 samples. The percentage of the oxygen vacancy concentration is found to be 23%, 33% for x = 0.10, 0.15, respectively, as calculated by the ratio of MB/(LB + MB + HB).
:Fe3+ is calculated as 33.3:66.7 and 39.9:60.1 for x = 0.10, 0.15 respectively. The Ca 2p spectra deconvoluted into two wide peaks of Ca 2p3/2 ∼ 346.26 eV and Ca 2p1/2 ∼ 349.81 eV for x = 0.10, 0.15 are shown in Fig. 6(c), which are mainly ascribed to Ca–O bonds.35 Fig. 6(d) shows the O 1s peak is de-convoluted into three peaks; the lower binding (LB) energy, medium binding (MB) energy and higher binding (HB) energy for x = 0.10, 0.15 samples. The percentage of the oxygen vacancy concentration is found to be 23%, 33% for x = 0.10, 0.15, respectively, as calculated by the ratio of MB/(LB + MB + HB).
 |
| | Fig. 6 Deconvoluted core level XPS spectra of (a) Bi 4f, (b) Fe 2p, (c) Ca 2p and (d) O 1s lines Bi1−xCaxFeO3 samples with x = 0.10 and 0.15. | |
3.5 Vibrating sample measurement (VSM) analysis
Magnetic measurements of prepared samples were recorded to study the effect of Ca doping on magnetic properties of BiFeO3 nanoparticles. The magnetic hysteresis (M–H) loops for Bi1−xCaxFeO3 samples are shown in Fig. 7(a). The antiferromagnetic (AFM) and weak ferromagnetic (WFM) M–H curves consists two parts that are paramagnetic (PM) and ferromagnetic (FM). In order to separate out ferromagnetic and paramagnetic contributions in M–H hysteresis loops, the M–H loops were analyzed by the following equation:
 |
| | Fig. 7 (a) Magnetization versus magnetic field (M–H) curves for Bi1−xCaxFeO3 nanoparticles. Paramagnetic and ferromagnetic fitted magnetization versus magnetic field (M–H) curve at room temperature for (b) x = 0.10, (c) x = 0.15 and (d) x = 0.20. (e) Canted and collinear anti-ferromagnetic spin structures in Ca doped Bi1−xCaxFeO3 through Dzyaloshinskii–Moriya interactions. | |
The first term demonstrates the ferromagnetic part and the second term represents the linear contributions from AFM and/or PM part.36,37 The fitted M–H curve along with FM and PM parts for Bi1−xCaxFeO3: x = 0.10, 0.15 and 0.20 samples are shown in Fig. 7(b)–(d) and the parameters obtained from fitting are listed in Table 1. The obtained parameters agree with the canted antiferromagnetic order of BiFeO3 ceramics and are comparable with the reported values.38,39 Park et al. demonstrated that the weak ferromagnetic response in BiFeO3 can be initiated when the size of the nanoparticles is less than 95 nm.9,38,39 The magnetic response increases rapidly with decreasing the size of the nanoparticles below 62 nm, the period length of spiral spin structure. TEM measurements confirmed that the size of Ca doped BiFeO3 nanoparticles vary in the range 45–90 nm which is responsible for the observation of weak ferromagnetism in present samples. The antiferromagnetic materials contain two spin sublattices, with ferromagnetic interaction within each sublattice and antiferromagnetic interaction in the inter-sublattices. This two sublattices model leads to long-range collinear antiparallel spin structure with zero net magnetic moment due to complete spin compensation between these two sublattices. Moreover, incomplete spin compensation between these two sublattices can also occur in antiferromagnetic materials.9 In nano-sized antiferromagnetic materials, long-range antiferromagnetic ordering often gets disturbed at the particle surface. Hence, in antiferromagnetic materials, if resultant small magnetic moment is observed, it could be due to the presence of uncompensated spins in the two sublattices. In addition, the higher surface-to-volume ratio in nanoparticles results in uncompensated spins at the surface and enhanced magnetization (due to disturbed antiferromagnetic ordering). It can be observed from the fitted MH loops that the antiferromagnetic contribution increases continuously with increasing Ca concentration up to x = 0.20 samples. However, the ferromagnetic ordering contribution increases with increasing Ca concentration up to x = 0.15 samples and then decreases significantly for x = 0.20 sample. The decrease of ferromagnetic ordering component in x = 0.20 sample is attributed to the increasing percentage of Pnma symmetry with increasing Ca concentration which favors the collinear antiferromagnetic ordering instead of canted spiral spin ordering. Moreover, Dzyaloshinskii–Moriya explained ‘ferroelectrically-induced ferromagnetism’, the fundamental idea of magnetization in antiferromagnetic materials due to the modification in spins structure as a result of the ferroelectric distortions.40,41 Therefore, it can be suggested that the improved magnetic ordering in Ca doped BiFeO3 nanoparticles is due to the modifications in its ‘canted’ spin structure and it leads to the enhanced magnetic properties up to x = 0.15 samples and after that canted AFM ordering changing to collinear antiferromagnetic ordering x = 0.20 sample as illustrated in Fig. 7(e). These canted spin antiferromagnetic ordering and collinear antiferromagnetic ordering in Ca doped BiFeO3 nanoparticles are further confirmed in EPR studies.
Table 1 Parameters extracted from fitting the magnetization hysteresis loops of Bi1−xCaxFeO3 nanoparticles
| S. No. |
Sample |
Paramagnetic contribution |
Ferromagnetic contribution |
| χ × 10−6 emu g−1 gauss−1 |
Hci (gauss) |
MSFM (emu g−1) |
MRFM (emu g−1) |
| 1 |
x = 0.00 |
6.35 |
1300 |
0.0099 |
0.005 |
| 2 |
x = 0.05 |
8.75 |
1200 |
0.018 |
0.0053 |
| 3 |
x = 0.10 |
7.0 |
640 |
0.045 |
0.008 |
| 4 |
x = 0.15 |
7.25 |
300 |
0.09 |
0.01 |
| 5 |
x = 0.20 |
12.9 |
275 |
0.0235 |
0.0025 |
3.6 Electron paramagnet resonance (EPR) analysis
Fig. 8(a) shows the room temperature EPR spectra of Ca doped BiFeO3 nanoparticles. The three parameters (g-factor, asymmetry parameter Pasy and signal width ΔBp–p) calculated from EPR measurements have been correlated with the magnetic structure of the samples. The g values were obtained by using the formula hν = gμBH, where ν is operating frequency, μB is Bohr magneton and h is Planck's constant. The asymmetry parameter Pasy is defined as Pasy = (1 − hU/hL) where hU is the height of the absorption peak above the base line and hL is the height of the absorption peak below the base line of the first derivative of the magnetic resonance absorption signal. ΔBp–p is the width of the signal defined as the separation between the upper peak and the lower peak.42,43 Calculated values of g, Pasy and ΔBp–p are given in Table 2. Interestingly, the EPR line shifts towards lower magnetic field which gives increasing g values for x = 0.0 to 0.15 samples. However, for x = 0.20 sample, the EPR line shifts towards higher magnetic field which results in lower g value. These EPR results suggest two different spin structures for x < 0.15 and x > 0.15 sample. The degree of spin canting may be calculated through vector parameter D which is defined as: D ≈ (Δg/g)Jsuper, where Δg is deviation of g from value 2, Jsuper is the superexchange interaction coefficient and is assumed to be constant for all the samples. The value D is largest for x = 0.15 sample as seen from Δg/g values in Table 2. This supports the M–H loop measurement results that the ferromagnetic interactions are stronger in x = 0.15 sample as compared to the other samples. The high asymmetry in the EPR spectra suggested the EPR signal is composed of two peaks. For the sake of convenience, the broad and narrow signals are designated as signal A and signal B, respectively as shown in Fig. 8(b)–(d) for Bi1−xCaxFeO3 (x = 0.0, 0.15 and 0.20) samples. The broad and weak Gaussian signal A with Hpp = 2236 gauss (peak-to-peak line width) and g-value as 2.19 and the intense and narrow Gaussian signal B with Hpp = 1194 gauss and g-value as 2.04 were fitted for Bi1−xCaxFeO3 (x = 0.0, 0.15 and 0.20) samples, shown in Fig. 8(b)–(d). The signal A is related to the ferromagnetic part and the signal B related to the paramagnetic part.44 With increasing the Ca concentration up to the 15%, the area under the signal A increases, confirming the enhanced ferromagnetism in these samples. There is a striking change in the EPR spectrum of 20% Ca doped BiFeO3 sample, where the paramagnetic signal B converges to sharp and almost symmetric peak having minimum line width which indicates a substantial change in the magnetic environment for Fe3+ ion. These results demonstrate an induced phase transition from an incommensurately cycloidal modulated state to one with nearly homogeneous (collinear) spin order. This type of magnetic phase transformation has also been reported by Kumar et al.37
 |
| | Fig. 8 (a) Electrons paramagnetic resonance (EPR) spectra of Bi1−xCaxFeO3 nanoparticles at room temperature. Fitted EPR spectra of (b) x = 0.0, (c) x = 0.15 and (d) x = 0.20. | |
Table 2 Calculated EPR parameters of Bi1−xCaxFeO3 nanoparticles at room temperature
| S. No. |
Sample |
g |
Δg/g |
ΔBp–p (gauss) |
Pasy |
| 1 |
x = 0.0 |
2.05 |
0.028 |
1098 |
0.22 |
| 2 |
x = 0.05 |
2.11 |
0.056 |
987.8 |
0.7 |
| 3 |
x = 0.10 |
2.13 |
0.067 |
1072 |
0.47 |
| 4 |
x = 0.15 |
2.19 |
0.095 |
904.4 |
0.31 |
| 5 |
x = 0.20 |
2.004 |
0.002 |
287.5 |
0.10 |
3.7 Mössbauer spectroscopy
The electronic and magnetic properties of Bi1−xCaxFeO3 nanoparticles with x = 0, 0.05, 0.10 and 0.15 are also systematically investigated by Mössbauer spectroscopy. The Mössbauer spectra provide information about the electronic density at the nuclei (through isomer shift IS), the possible electric field gradient (quadrupole splitting QS) and the magnetic environment (magnetic hyperfine splitting HF). Mössbauer spectra fitted with one doublet and one sextet for pure BiFeO3 nanoparticles and fitted with one sextet for Bi1−xCaxFeO3 nanoparticles with x = 0.05, 0.10 and 0.15 are shown in Fig. 9(a)–(d). In case of pure BiFeO3 nanoparticles, the Mössbauer spectrum contains a superposition of quadrupolar doublet and magnetic sextet as shown in Fig. 9(a). This collapsed quadrupolar (doublet and sextet) spectrum is related to the particle size distribution within the synthesized sample. Thes tiny doublet in Mössbauer spectrum may be related to superparamagnetic relaxation of smaller particles. The similar type of Mössbauer spectra is observed for BiFeO3 nanoparticles less than 95 nm as reported by Park et al.9 One sextet fitting hyperfine parameters are tabulated in Table 3. The isomer shift – IS which is a measure of the oxidation state of Fe atoms is found to be ∼0.37 mm s−1 for all the samples. We note that the values of IS are reported to be in the range of 0.6–1.7 mm s−1 for Fe2+; 0.05–0.5 mm s−1 for Fe3+ and −0.15 to 0.05 for Fe4+.45 This indicates that Fe3+ oxidation state is dominant in present samples. The QS is 0.1275, 0.0977, 0.0259 and 0.0195 mm s−1 for Bi1−xCaxFeO3: x = 0.0, 0.05, 0.10 and 0.15. The decrease in QS with increasing Ca content in BiFeO3 can be explained by compressive distortion of oxygen octahedron and/or displacement of Fe ions across diagonal of pseudocubic crystal cell. Hyperfine magnetic field HF increases from 47.374–49.46 tesla with increasing Ca content from 5–15% in BiFeO3 samples. It is known that Fe3+ ions do not possess any orbital moment and as a consequence there is no orbital magnetism. Therefore, hyperfine magnetic field is only caused by contact Fermi interaction, which is proportional to the difference of electronic densities belonging to spin-up and spin-down states. The difference appears due to polarization of the 3d electronic shell caused by interaction of Fe ion with neighbor oxygen ions. From the Rietveld analysis, the lattice parameter decreases with increasing Ca concentration in BiFeO3 samples and Ca2+ substitution at Bi3+ site leads to the oxygen vacancies. Hence, the distortion in the crystal structure with increasing Ca2+ content results in the change of Fe ion surrounding which has lead to the hyperfine magnetic structure. Lebeugle et al. reported that the spin cycloid causes the broadening of Mössbauer spectra line.46 The line broadening (FWHM: Full Width at Half Maximum) of the sextet decreases with increasing Ca content in BiFeO3 samples and this may be associated with the destruction of the spin cycloid in BiFeO3. The similar type of effects has already been reported in Pr doped BiFeO3.47 The larger FWHM and asymmetry of intensity of the lines suggested the existence of at least two nonequivalent positions of Fe ions as shown in Fig. 10. The two-sextet fit matches reasonably well with experimental data points and the hyperfine parameters are presented in Table 4. A comparison of the obtained values of the room temperature parameters of hyperfine interactions with data available in the literature for iron ions with different oxygen co-ordinations indicates that two sextets with approximately equal shift of the line (IS ≈ 0.37–0.39) and small QS correspond to Fe3+ ions in a weakly distorted octahedral oxygen environment (Fe–O). The quadrupole splitting and hyperfine magnetic fields of the two sextets approach to each other with increasing Ca concentration in BiFeO3 samples and this indicates the appearance of equivalence of Fe ions.
 |
| | Fig. 9 Room temperature Mössbauer spectra for (a) x = 0, (b) x = 0.05, (c) x = 0.10 and (d) x = 0.15 samples fitted with one sextet. | |
Table 3 Hyperfine parameters obtained from one-sextet fitting of the Mössbauer spectra: IS—Isomer Shift, QS—Electric Quadrupole Splitting, HF—hyperfine magnetic field of Bi1−xCaxFeO3 nanoparticles
| Sample |
Type |
HF (tesla) |
QS (mm s−1) |
IS (mm s−1) |
Area ratio |
| x = 0.0 |
Sextet 1 |
49.70 |
0.1275 |
0.377 |
82.01% |
| Doublet |
|
0.9069 |
0.1918 |
17.99% |
| x = 0.05 |
Sextet 1 |
47.374 |
0.0977 |
0.3757 |
100% |
| x = 0.10 |
Sextet 1 |
49.261 |
0.0259 |
0.3789 |
100% |
| x = 0.15 |
Sextet 1 |
49.46 |
0.0195 |
0.3735 |
100% |
 |
| | Fig. 10 Mössbauer spectra for (a) x = 0, (b) x = 0.05, (c) x = 0.10 and (d) x = 0.15 samples fitted with two sextets. | |
Table 4 Hyperfine parameters obtained from two-sextet fitting of the Mössbauer spectra: IS—Isomer Shift, QS—Electric Quadrupole Splitting, HF—hyperfine magnetic field of Bi1−xCaxFeO3 nanoparticles
| Sample |
Type |
HF (tesla) |
QS (mm s−1) |
IS (mm s−1) |
Area ratio (%) |
| x = 0.0 |
Sextet 1 |
48.30 |
0.0718 |
0.3856 |
25.44 |
| Sextet 2 |
50.23 |
0.1456 |
0.3779 |
56.87 |
| Doublet |
|
0.7886 |
0.2149 |
17.69 |
| x = 0.05 |
Sextet 1 |
47.39 |
0.0754 |
0.3964 |
37.74 |
| Sextet 2 |
49.62 |
0.1009 |
0.3755 |
62.26 |
| x = 0.10 |
Sextet 1 |
47.17 |
0.0049 |
0.3957 |
35.08 |
| Sextet 2 |
49.48 |
0.0382 |
0.374 |
64.92 |
| x = 0.15 |
Sextet 1 |
47.19 |
0.009 |
0.3672 |
32.41 |
| Sextet 2 |
49.41 |
0.022 |
0.3736 |
67.59 |
4. Conclusions
In summary, Bi1−xCaxFeO3 nanoparticles have been successfully synthesized by the sol–gel route. Substitution induced structural and magnetic phase transitions have been studied in detail. XRD results showed that the contribution of orthorhombic phase increases with increasing Ca content in BiFeO3 nanoparticles. The TEM images demonstrated that particle size varies in nano range (45–90 nm) and decreases with increasing Ca concentration in BiFeO3. A significant change in intensity, position and width of the E(LO1), E(TO2), E(LO9) Raman modes of Ca substituted BiFeO3 samples revealed the structural transition from rhombohedral symmetry (space group R3c) to orthorhombic (Pnma). XPS spectra indicated the dominancy of Fe3+ oxidation state and slight increase in oxygen vacancies with increasing Ca content in BiFeO3 samples. The ferromagnetic ordering component increases with increasing Ca content and it is maximum for 15% Ca doped BiFeO3 sample. However, the ferromagnetic ordering component again decreases significantly for 20% of Ca doping due to collinear antiferromagnetic ordering in the Pnma phase for this sample, which is further confirmed from EPR studies. Local magnetic behavior investigated by 57Fe Mössbauer spectroscopy rules out any valence fluctuations of Fe with Ca doping and the hyperfine field corroborates the magnetization data. One sextet Mössbauer spectra fitting have showed systematic decrease in QS hyperfine parameter and line-width with increasing Ca content in BiFeO3 nanoparticles suggesting the destruction of spin cycloid. The analysis of the spectral asymmetry demonstrated the line broadening mechanism and fitting with two sextets give further evidence for spin cycloid destruction.
Acknowledgements
Authors are thankful to Department of science and Technology (DST) (Grant number SR/FTP/PS-91/2009) to carry out this research work. The authors are thankful to SAIF AIIMS, New Delhi and SAIF IIT, Chennai for TEM, VSM and EPR measurements. The authors are also thankful to INUP program IISc, Bangalore for Raman measurement and XPS measurements.
Notes and references
- M. Fiebig, T. Lottermoser, D. Frohlich, A. V. Goltsev and R. V. Pisarev, Nature, 2002, 419, 818 CrossRef CAS PubMed.
- N. A. Hill, J. Phys. Chem., 2000, 104, 6694 CrossRef CAS.
- Y. Li, M.-S. Cao, D.-W. Wang and J. Yuan, RSC Adv., 2015, 5, 77184 RSC.
- B. Sun, Y. Liu, W. Zhao and P. Chen, RSC Adv., 2015, 5, 13513 RSC.
- S. Das, S. Rana, S. M. Mursalin, P. Rana and A. Sen, Sens. Actuators, B, 2015, 218, 122 CrossRef CAS.
- L. W. Martin, Dalton Trans., 2010, 39, 10813 RSC.
- G. Catalan and J. F. Scott, Adv. Mater., 2009, 21, 2463 CrossRef CAS.
- M. Escobar Castillo, V. V. Shvartsman, D. Gobeljic, Y. Gao, J. Landers, H. Wende and D. C. Lupascu, Nanotechnology, 2013, 24, 355701 CrossRef CAS PubMed.
- T. J. Park, G. C. Papaefthymiou, A. J. Viescas, A. R. Moodenbaugh and S. S. Wong, Nano Lett., 2007, 7, 766 CrossRef CAS PubMed.
- B. Bhushan, A. Basumallick, S. K. Bandopadhyay, N. Y. Vasanthacharya and D. Das, J. Phys. D: Appl. Phys., 2009, 42(6), 065004 CrossRef.
- J. Zhao, X. Zhang, S. Liu, W. Zhang and Z. Liu, J. Alloys Compd., 2013, 557, 120 CrossRef CAS; Z. X. Cheng, X. L. Wang, Y. Du and S. X. Dou, J. Phys. D: Appl. Phys., 2010, 43, 242001 CrossRef; T. Karthik, T. Durga Rao, A. Srinivas and S. Asthana, J. Mater. Sci.: Mater. Electron., 2015, 26, 8676 CrossRef.
- K. Sardar, J. Hong, G. Catalan, P. K. Biswas, M. R. Lees, R. I. Walton, J. F. Scott and S. A. T. Redfern, J. Phys.: Condens. Matter, 2012, 24, 045905 CrossRef PubMed.
- J. A. Schiemer, R. L. Withers, Y. Liu and M. A. Carpenter, Chem. Mater., 2013, 25, 4436 CrossRef CAS PubMed.
- X. Wang, S. Y. Wang, W. F. Liu, X. J. Xi, H. Zhang, F. Guo, X. L. Xu, M. Li, L. Liu, C. Zhang, X. Li and J. B. Yang, J. Nanopart. Res., 2015, 17, 209, DOI:10.1007/s11051-015-3018-1.
- P. Tirupathi and A. Chandra, J. Alloys Compd., 2013, 564, 151 CrossRef CAS; A. T. Kozakov, A. G. Kochur, V. I. Torgashev, K. A. Googlev, S. P. Kubrin, V. G. Trotsenko, A. A. Bush and A. V. Nikolskii, J. Alloys Compd., 2016, 664, 392 CrossRef.
- W. Chen, A. J. Williams, L. Ortega-San-Martin, M. Li, D. C. Sinclair, W. Zhou and J. P. Attfield, Chem. Mater., 2009, 21, 2085 CrossRef CAS.
- J. Schiemer, R. Withers, L. Norén, Y. Liu, L. Bourgeois and G. Stewart, Chem. Mater., 2009, 21(18), 4223 CrossRef CAS.
- A. G. Gavriliuk, V. V. Struzhkin, L. S. Lyubutin, S. G. Ovchinnikov, M. Y. Hu and P. Chow, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155112 CrossRef.
- G. Catalan, K. Sardar, N. S. Church, J. F. Scott, R. J. Harrison and S. A. T. Redfern, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 212415 CrossRef.
- X. Wang, S. Y. Wang, W. F. Liu, F. Guo, X. J. Xi, H. J. Wang and D. J. Li, Mod. Phys. Lett. B, 2014, 28(7), 1450050 CrossRef.
- S. Gosh, S. Das Gupta, A. Sen and H. S. Maiti, J. Am. Ceram. Soc., 2005, 88(5), 1349–1352 CrossRef CAS; M. Arora, P. C. Sati, S. Chauhan, S. Chhoker, A. K. Panwar and M. Kumar, J. Supercond. Novel Magn., 2013, 26, 443 CrossRef.
- S. M. Selbach, T. Tybell, M. A. Einarsrud and T. Grande, Chem. Mater., 2007, 19, 6478 CrossRef CAS; S. Chauhan, M. Kumar, S. Chhoker and S. C. Katyal, J. Alloys Compd., 2016, 666, 454 CrossRef.
- J. Rodriguez-Carvajal, FullProf: A Rietveld Refinement and Pattern Matching Analysis Program (Version: April 2008), Laboratoire Léon Brillouin (CEA-CNRS), France, 2000 Search PubMed.
- P. Hermet, M. Goffinet, J. Kreisel and P. Ghosez, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 220102 CrossRef.
- P. Chen, X. Xu, C. Koenigsmann, A. C. Santulli, S. S. Wong and J. L. Musfeldt, Nano Lett., 2010, 10, 4526 CrossRef CAS PubMed.
- A. A. Porporati, K. Tsuji, M. Valant, A. K. Axelsson and G. Pezzotti, J. Raman Spectrosc., 2010, 41, 84 CrossRef CAS.
- J. Bielecki, P. Svedlindh, D. T. Tibebu, S. Cai, S.-G. Eriksson, L. Börjesson and C. S. Knee, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 184422 CrossRef.
- N. Suzuki and H. Kamimura, J. Phys. Soc. Jpn., 1973, 35, 985 CrossRef CAS.
- J.-P. Zhou, R.-J. Xiao, Y.-X. Zhang, Z. Shi and G.-Q. Zhu, J. Mater. Chem. C, 2015, 3, 6924 RSC.
- M. Cazayous, D. Malka, D. Lebeugle and D. Colson, Appl. Phys. Lett., 2007, 91, 071910 CrossRef.
- J. Hlinka, J. Pokorny, S. Karimi and I. M. Reaney, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 020101 CrossRef.
- M. O. Ramirez, M. Krishnamurthi, S. Denev, A. Kumar, S. Y. Yang, Y. H. Chu, E. Saiz, J. Seidel, A. P. Pyatakov, A. Bush, D. Viehland, J. Orenstein, R. Ramesh and V. Gopalan, Appl. Phys. Lett., 2008, 92, 022511 CrossRef.
- Z. Quan, W. Liu, H. Hu, S. Xu, B. Sebo, G. Fang, M. Li and X. Zhao, J. Appl. Phys., 2008, 104, 084106 CrossRef.
- A. Tamilselvan, S. Balakumar, M. Sakar, C. Nayek, P. Murugavel and K. S. Kumar, Dalton Trans., 2014, 43, 5731 RSC.
- B. Demri and D. Muster, J. Mater. Process. Technol., 1995, 55, 311 CrossRef.
- L. R. Shah, H. Zhu, W. G. Wang, B. Ali, T. Zhu, X. Fan, Y. Q. Song, Q. Y. Wen, H. W. Zhang, S. I. Shah and J. Q. Xiao, J. Phys. D: Appl. Phys., 2010, 43, 035002 CrossRef.
- P. Kumar, N. Shankhwar, A. Srinivasan and M. Kar, J. Appl. Phys., 2015, 117, 194103 CrossRef.
- F. Huang, Z. Wang, X. Lu, J. Zhang, K. Min, W. Lin, R. Ti, T. T. Xu, J. He, C. Yue and J. Zhu, Sci. Rep., 2013, 3, 2907 Search PubMed.
- S.-Z. Lu and X. Qi, J. Am. Ceram. Soc., 2014, 97(7), 2185 CrossRef CAS.
- H. Feng, J. Magn. Magn. Mater., 2010, 322, 1765 CrossRef CAS.
- M. Sakar, S. Balakumar, P. Saravanan and S. Bharathkuma, Nanoscale, 2015, 7, 10667 RSC.
- D. Maurya, H. Thota, A. Garg, B. Pandey, P. Chand and H. C. Verma, J. Phys.: Condens. Matter, 2009, 21, 026007 CrossRef PubMed.
- M. Arora, P. C. Sati, S. Chauhan, M. Kumar and S. Chhoker, Mater. Lett., 2014, 132, 327 CrossRef CAS.
- D. Karmakar, S. K. Mandal, R. M. Kadam, P. L. Paulose, A. K. Rajarajan, T. K. Nath, A. K. Das, I. Dasgupta and G. P. Das, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 144404 CrossRef.
- N. N. Greenwood and T. C. Gibb, Mössbauer Spectroscopy, Chapman and Hall, London, 1971 Search PubMed.
- D. Lebeugle, D. Colson, A. Forget, M. Viret, P. Bonville, J. F. Marucco and S. Fusil, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024116 CrossRef.
- R. Xiao, V. O. Pelenovich and D. Fu, Appl. Phys. Lett., 2013, 103, 012901 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02316a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 



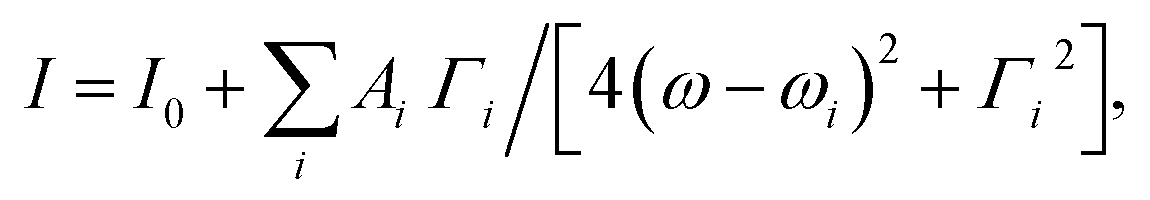 where i is the peak number, I0 accounts for the background intensity, ωi is the center frequency, Γi is the full width at half maxima (FWHM), and Ai is the area of ith peak. The fitted spectra for Bi1−xCaxFeO3 (x = 0.0, 0.15 and 0.20) nanoparticles are shown in Fig. 4(b)–(d), respectively. The observed modes position and FWHM of E and A modes for Bi1−xCaxFeO3 (x = 0–0.20) nanoparticles are summarized in ESI Table S2.† First Raman active mode near 75 cm−1 splits into two modes E(TO1) and E(LO1). Similarly both LO and TO modes are present for E(8) and E(9). The Raman modes doublet E(TO1), E(LO1) near 75 cm−1, E(TO2) mode at 139.2 cm−1 and A1(TO1) mode at 172.3 cm−1 are due to the displacement of Bi atoms. This displacement is caused by the activation of lone pair 6s2 electrons of Bi3+ along the c-axis of hexagonal unit cell. The Raman modes E(TO1)/E(LO1) provide the key information about the magnetoelectric coupling in BiFeO3 due to its strong polar character and further regulates the dielectric constant of the material. The shifting of the first order modes E(LO1)/E(TO1) with increasing the Ca content in BiFeO3 samples is associated with strain via phonon deformation potentials α, β and γ,
where i is the peak number, I0 accounts for the background intensity, ωi is the center frequency, Γi is the full width at half maxima (FWHM), and Ai is the area of ith peak. The fitted spectra for Bi1−xCaxFeO3 (x = 0.0, 0.15 and 0.20) nanoparticles are shown in Fig. 4(b)–(d), respectively. The observed modes position and FWHM of E and A modes for Bi1−xCaxFeO3 (x = 0–0.20) nanoparticles are summarized in ESI Table S2.† First Raman active mode near 75 cm−1 splits into two modes E(TO1) and E(LO1). Similarly both LO and TO modes are present for E(8) and E(9). The Raman modes doublet E(TO1), E(LO1) near 75 cm−1, E(TO2) mode at 139.2 cm−1 and A1(TO1) mode at 172.3 cm−1 are due to the displacement of Bi atoms. This displacement is caused by the activation of lone pair 6s2 electrons of Bi3+ along the c-axis of hexagonal unit cell. The Raman modes E(TO1)/E(LO1) provide the key information about the magnetoelectric coupling in BiFeO3 due to its strong polar character and further regulates the dielectric constant of the material. The shifting of the first order modes E(LO1)/E(TO1) with increasing the Ca content in BiFeO3 samples is associated with strain via phonon deformation potentials α, β and γ,





