
Nanostructured double hydrophobic poly(styrene-b-methyl methacrylate) block copolymer membrane manufactured via a phase inversion technique†
Abstract
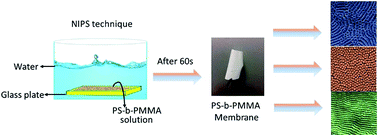
In this paper, we demonstrate the formation of nanostructured double hydrophobic poly(styrene-b-methyl methacrylate) (PS-b-PMMA) block copolymer membranes via a state-of-the-art phase inversion technique. The nanostructured membrane morphologies are tuned by different solvent and block copolymer compositions. The membrane morphology has been investigated using FESEM, AFM and TEM. Morphological investigation shows the formation of both cylindrical and lamellar structures on the top surface of the block copolymer membranes. The PS-b-PMMA, with an equal block length (PS160 K-b-PMMA160 K), exhibits both cylindrical and lamellar structures on the top layer of the asymmetric membrane. All membranes fabricated from PS160 K-b-PMMA160 K show incomplete pore formation in both cylindrical and lamellar morphologies during the phase inversion process. However, the PS-b-PMMA (PS135 K-b-PMMA19.5 K) block copolymer, with a short PMMA block, allowed us to produce open pore structures with ordered hexagonal cylindrical pores during the phase inversion process. The resulting PS-b-PMMA nanostructured block copolymer membranes have pure water flux from 105–820 L m−2 h− bar− and 95% retention of PEG50 K.
 Please wait while we load your content...
Please wait while we load your content...

