
Photoresponsive assemblies of linear-dendritic copolymers containing azobenzene in the dendron interior: the effect of the dendron structure on dye encapsulation and release†
Nagendra Kalvaab,
Nitin B. Basutkara and
Ashootosh V. Ambade*ab
aPolymer Science and Engineering Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune-411008, India. E-mail: av.ambade@ncl.res.in
bAcademy of Scientific and Innovative Research, New Delhi, India
First published on 26th April 2016
Abstract
Hydrophobic dendrons with different numbers and positions of azobenzenes as well as different groups – benzyl and dodecyl, on the periphery were synthesised and attached to poly(ethylene glycol) using copper-catalysed azide–alkyne cycloaddition to obtain linear-dendritic copolymers. Self-assembly of the polymers in aqueous solution was characterised using dynamic light scattering (DLS), transmission electron microscopy (TEM) and critical micelle concentration (cmc). Formation of H-aggregates during micellisation was shown for polymers with a higher number of azobenzene units. Photoisomerisation of azobenzene in the assemblies was studied and the rate constant of thermal photoisomerisation was calculated. Release of hydrophobic dye Nile red upon photoisomerisation of azobenzene occurred without disruption of micellar aggregates. Dye release varied with the pathway – thermal or visible light irradiation, followed for cis–trans isomerisation. The encapsulation capacity of the micelles and extent of dye release in either pathway were found to be influenced by the dendron structure. A polymer with a lower number of azobenzenes and aliphatic periphery on the dendron showed significantly different behaviour than polymers with a larger number of aromatic units.
Introduction
Photoresponsive polymers are an important class of smart materials due to their potential applications in various fields including liquid crystal displays,1–3 optical data storage,4,5 and as drug delivery systems.6–9 Light as a stimulus can be triggered at a specific position easily and precisely at a desired time that allows the properties of photoresponsive polymers like conformation and polarity to be reversibly altered. This is achieved through incorporation of chromophores such as azobenzene that show geometrical isomerism or dithienylethene and spiropyran that switch between ring opening and closing.10,11 The change in polarity or conformation of the polymer may cause release of the encapsulated guest molecule hence, photoresponsive polymer assemblies are potential candidates for controlled release systems.12,13Azobenzene is one of the most widely studied chromophore by polymer chemists14 due to its fast and reversible photoisomerisation. The trans isomer is converted to cis isomer upon exposure to UV light and the process is reversed upon illumination with visible light or thermally by storage in the dark. cis-Isomer of azobenzene is more polar due to higher dipole moment and hence the photoisomerisation leads to imbalance in hydrophilic/hydrophobic ratio of the polymer. This can be exploited to cause complete disruption or changes in morphology of polymer assemblies leading to release of the guest molecules.15,16 On the other hand, the imbalance may not be enough to cause disassembly or morphology change of the aggregate but the encapsulated dye molecule may be released due to change in polarity of its microenvironment.17 Further modulation of dye release is possible by subjecting the polymer to a series of trans–cis–trans photoisomerisation cycles with intermittent thermal restoration period as shown recently for micelles from a polymer with azobenzene in the side chain.18
Azobenzene containing dendritic polymers, particularly dendrimers and hyperbranched polymers have also been studied as compact nanoscale photoresponsive macromolecules.19–21 Linear-dendritic copolymers22–24 are an interesting class of polymer architecture that combines useful attributes of the perfectly branched dendron and linear polymer chain, for example defined number of functionalities can be installed at desired positions in the dendron. Incorporation of light-responsive moieties has been less explored in linear-dendritic copolymers compared to linear polymers. Azobenzene containing amphiphilic linear-dendritic copolymers with 4-cyanoazobenzene on dendron periphery were shown to self-assemble in water into morphologies such as cylindrical micelles, sheet-like micelles, tubular micelles and polymersomes with increasing dendron generation.25 Later, same copolymers with 4-isobutyloxyazobenzene on periphery were shown to form stable vesicles in water that displayed significant membrane deformation and subsequent dye release upon UV light irradiation.26 Dye release could be tuned by varying the content of 4-isobutyloxyazobenzene on periphery by optimizing free volume available for isomerisation.27 In these reports, cis-to-trans isomerisation was effected by incubating the sample in dark.
Photoisomerisation studies on dendrimers with multiple azobenzenes in the interior have shown that the same dendrimer forms several distinct states that possess different polarity and hydrodynamic volumes due to difference in the extent of trans and cis isomers.28,29 The studies were conducted in organic solvent since dendrimers were not amphiphilic. We hypothesised that incorporation of azobenzene units in the dendron interior of an amphiphilic linear-dendritic copolymer would afford better control over tuning the polarity of micellar core using photoirradiation. Thus, it may be possible to build stable polymer assemblies and exercise fine control over the dye release by varying the position and number of azobenzenes in the dendron. Furthermore, variation in the number and position of azobenzene can also be accomplished within dendrons of the same generation. This structural variation is likely to affect the volume of dendron interior and packing efficiency, thereby influencing the dye encapsulation capacity and morphology. Here, we describe design and synthesis of amphiphilic linear-dendritic copolymers with variation in position of azobenzene in the dendron. To gain further insight into the effect of dendron structure, different hydrophobic groups were also installed on the dendron periphery. Photoinduced dye release from copolymer micelles during azobenzene isomerisation cycle was found to depend on number of azobenzene units and the nature of hydrophobic groups. cis–trans isomerisation was carried out by photo as well as thermal pathway and the extent of dye release in the two processes was compared.
Experimental
Materials
3,5-Dihydroxy benzyl alcohol, 4-aminobenzyl alcohol, methoxy-PEG (Mn = 2000 g mol−1, PDI = 1.1), dodecyl bromide, copper(I) bromide, PMDETA, propargyl bromide, lithium aluminium hydride and Nile red were purchased from Aldrich Chemicals Co. Benzyl bromide, triphenyl phosphine, carbon tetrabromide, 18-crown-6 ether, phenol, sodium azide, potassium carbonate and triethylamine were purchased from Avra Chemicals, Spectrochem and Merck Chemicals, India. Dichloromethane was dried over CaH2 and acetone was dried over K2CO3, distilled and stored in a Schlenk flask. Tetrahydrofuran (THF) was passed through alumina and dried on sodium wire and freshly distilled when required. All compounds were purified using column chromatography on silica gel (mesh size 60–120 and 220–440).Instrumentation
1H NMR was recorded on Bruker spectrometer (200 MHz or 400 MHz). FT-IR spectra were recorded on Perkin Elmer FT-IR spectrum GX instrument by making KBr pellets. Gel permeation chromatography (GPC) analysis was performed using Viscotek instrument comprising VE 1122 pump, Viscotek VE 3580 RI detector, and Viscotek VE 3210 UV-vis detector in THF or using Viscotek PL-GPC-220 instrument in CHCl3 using polystyrene standards. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) spectra were recorded on an ABSCIEX TOF/TOF 5800 mass spectrometer using dithranol as matrix. Dynamic light scattering experiments were performed on 90 Plus particle size analyser by Brookhaven Instruments Corporation at an angle of 90° equipped with laser beam of 633 nm wavelength. Transmission electron microscopy images were taken on FEI Technai T20 instrument operating at 200 kV. UV-vis spectra were recorded on Specord 210 plus Analytikjena spectrophotometer. Fluorescence emission spectra were recorded on CARY Eclipse spectrometer. Photoisomerization was performed using Philips TUV 8 W lamp for 365 nm and Naava LT 6 W T5 lamp for 450 nm irradiation. For dye release studies, a 100 W UV lamp equipped with 365 nm filter (intensity 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μW cm−2) was used.
:2) was added the azide-PEG2000 and resultant mixture was degassed for 15 min. CuBr (1 eq.) and PMDETA (1 eq.) were then added and allowed to stir for 24 h under argon atmosphere. The progress of the reaction was monitored by the disappearance of azide stretching frequency at 2105 cm−1. Organic layer was concentrated under reduced pressure. Copper salts were removed by passing through neutral alumina. Crude product was purified by precipitating in cold diethyl ether. The obtained orange-coloured solid was dried under high vacuum. Yield 65%.
000 μW cm−2) was used.
:2) was added the azide-PEG2000 and resultant mixture was degassed for 15 min. CuBr (1 eq.) and PMDETA (1 eq.) were then added and allowed to stir for 24 h under argon atmosphere. The progress of the reaction was monitored by the disappearance of azide stretching frequency at 2105 cm−1. Organic layer was concentrated under reduced pressure. Copper salts were removed by passing through neutral alumina. Crude product was purified by precipitating in cold diethyl ether. The obtained orange-coloured solid was dried under high vacuum. Yield 65%.
Polymer P1. 1H NMR (400.13 MHz, CDCl3) δ: 3.38 (s, 3H), 3.64 (bs, 174H), 4.52–4.55 (m, 4H), 4.68 (s, 2H, –CH2), 5.04–5.12 (m, 16H, –CH2), 6.58–6.70 (m, 9H, Ar H), 7.04 (d, 4H, J = 12 Hz), 7.30–7.42 (m, 20H, Ar H), 7.53 (d, 4H, J = 8 Hz), 7.74 (s, 1H, triazole), 7.88–7.91 (m, 8H, Ar H) ppm. 13C NMR (CDCl3, 50.32 MHz): 50.32, 59.11, 63.76, 69.55, 69.74, 70.24, 70.66, 72.04, 72.31, 101.61, 106.44, 106.99, 114.78, 115.24, 122.89, 123.90, 124.86, 127.61, 128.68, 136.82, 138.98, 139.33, 140.69, 144.94, 147.25, 152.49, 160.02, 160.33, 161.23 ppm. MALDI-TOF MS: calcd 3230, found 3218.
Polymer P2. 1H NMR (400.13 MHz, CDCl3) δ: 0.88 (t, 12H, J = 8 Hz), 1.26 (bs, 72H), 1.77 (m, 8H), 3.38 (s, 8H), 3.64 (bs, 172H), 3.96 (t, 8H, J = 8 Hz), 4.55 (m, 4H), 4.68 (s, 2H, –ArCH2), 5.07–5.12 (m, 8H), 6.42 (s, 2H, Ar H), 6.58–6.66 (m, 7H), 7.07 (d, 4H, J = 8 Hz), 7.54 (d, 4H, J = 8 Hz), 7.75 (s, 1H, triazole), 7.88–7.92 (m, 8H, Ar H) ppm. 13C NMR (CDCl3, 50.32 MHz): 14.23, 22.78, 26.14, 29.34, 29.44, 29.73, 32.01, 59.13, 62.98, 63.62, 64.28, 68.20, 70.66, 105.76, 115.23, 122.90, 124.87, 128.09, 138.68, 160.69 ppm. MALDI-TOF MS: calcd 3542, found 3549.
Polymer P3. 1H NMR (400.13 MHz, CDCl3) δ: 0.88 (t, 12H, J = 8 Hz), 1.27–1.47 (m, 72H), 1.82 (m, 8H), 3.38 (s, 3H), 3.65 (bs, 172H), 4.03 (t, 8H, J = 8 Hz), 4.54 (m, 4H), 4.67 (s, 2H), 4.99 (s, 4H), 5.11 (s, 8H, –ArCH2), 6.52–6.70 (m, 9H), 6.98 (d, 8H, J = 8 Hz), 7.52 (d, 8H, J = 8 Hz), 7.73 (s, 1H, triazole), 7.86–7.89 (m, 16H, Ar–H) ppm. 13C NMR (CDCl3, 125.76 MHz): 14.22, 22.79, 26.12, 29.32, 29.45, 29.76, 32.02, 50.35, 59.12, 63.77, 68.51, 69.83, 70.68, 72.06, 72.40, 101.87, 106.61, 114.83, 122.87, 123.91, 124.89, 128.08, 139.13, 139.64, 146.97, 152.57, 160.15, 161.90 ppm.
Polymer P4. 1H NMR (400.13 MHz, CDCl3) δ: 0.88 (t, 24H, J = 8 Hz), 1.26 (bs, 144H), 1.76 (m, 16H), 3.38 (s, 3H), 3.65 (bs, 174H), 3.94 (t, 16H, J = 8 Hz), 4.52–4.54 (m, 4H), 4.67 (s, 2H), 4.99–5.11 (m, 20H), 6.41 (s, 4H), 6.52–6.71 (m, 17H), 7.05 (d, 8H, J = 12 Hz), 7.53 (t, 8H, J = 12 Hz), 7.73 (s, 1H), 7.87–7.91 (t, 16H, J = 8 Hz) ppm. 13C NMR (CDCl3, 100.61 MHz): 14.06, 22.62, 25.97, 29.17, 29.27, 29.53, 29.56, 31.84, 50.14, 58.97, 68.01, 70.49, 71.85, 100.78, 101.59, 105.57, 106.39, 106.73, 114.56, 115.03, 122.74, 124.70, 127.92, 138.49, 139.00, 139.40, 147.02, 152.31, 159.93, 160.50, 161.17 ppm.
Results and discussion
Synthesis
Hydrophobic dendrons with azobenzene units in the dendritic backbone were synthesised by the convergent approach for benzyl ether dendrons.30 Two dendrons comprising two azobenzene units in the interior and either benzyl group (D1) or dodecyl chains (D2) on the periphery were synthesised (Schemes S1 and S2, ESI†). To achieve variation in number of azobenzenes and number of alkyl chains and position of azobenzene, two more dendrons containing four azobenzene units with four and eight dodecyl chains on the periphery (D3 and D4) were synthesised. Dendron D3 contained dodecyloxy-azobenzene units in the peripheral layer of dendron while D4 contained azobenzenes in the dendron interior (Schemes S2 and S3, ESI†). Dendrons D1–D3 are second-generation (G2) whereas D4 is third-generation (G3) dendron. The substitution pattern on the azobenzene units was maintained in all the dendrons for a straightforward comparison. An alkyne functionality was installed at the focal point of dendrons in the last step. Alkyne-functionalised dendrons were characterised by 1H NMR, 13C NMR, and MALDI-TOF techniques to ascertain structural integrity (Fig. S1–S32†).Monomethoxy poly(ethylene glycol) (PEG) functionalised with azide at one end was attached to alkyne-functionalised azo-dendrons by using copper-catalysed azide–alkyne cycloaddition (CuAAC), an efficient coupling method for polymers.31,32 The obtained polymers P1–P4 were purified by precipitation in cold diethyl ether. Chemical structures of the polymers are shown in Chart 1. 1H NMR spectra revealed signals for protons of dendron and PEG as well as for the triazole ring proton (7.73 ppm) formed during the click reaction and absence of alkyne proton near 2.5 ppm as illustrated in Fig. 1a for P3 as an example.
 | ||
| Chart 1 Chemical structures of linear-dendritic copolymers. | ||
 | ||
| Fig. 1 (a) 200 MHz 1H NMR spectrum of P3 in CDCl3. (b) FT-IR spectra for the click reaction. (c) Overlay of GPC chromatograms of P1–P4 and corresponding dendrons D1–D4. | ||
Absence of azide group was confirmed from FT-IR spectra that showed complete disappearance of the band near 2100 cm−1 (Fig. 1b). Gel permeation chromatography (GPC) analysis revealed monomodal peaks that confirmed the purity of polymers (Fig. 1c). Polymers P1 and P2 were also characterised by MALDI-TOF analysis (Fig. S33 and S34†) however, the analysis was not satisfactory for P3 and P4 even with different matrices. Literature reports exist on unsuccessful MALDI-TOF analysis for linear-dendritic copolymers with azobenzene on the dendron periphery.33
Self-assembly in water
Aqueous solutions of the amphiphilic linear-dendritic copolymers were prepared by the dialysis method at a concentration of 0.1 wt%. Water was slowly added to a solution in THF to induce aggregation and the mixture was dialysed against water to remove THF completely. The micellar aggregates are expected to comprise hydrophobic azo-dendrons in the micellar core and PEG chains in the corona in water. Morphology of the aggregates was investigated by transmission electron microscopy (TEM) analysis that revealed spherical aggregates for P1 and P2. Aqueous solution of P1 showed micelles of 137 nm diameter while solution of P2 showed similar aggregates with size of 22 nm (average of 40 particles as measured from TEM images) (Fig. 2a and c). Polymers P3 and P4 did not show defined aggregates in TEM analysis, however when N,N-dimethylformamide (DMF) was used for preparing aqueous solutions defined morphologies were observed (Fig. S36†) since the organic co-solvent used during the assembly is known to play a role in directing the morphology.34 Nevertheless, aqueous solutions prepared from THF were used for all further characterisation and studies. Dynamic light scattering (DLS) analysis showed that average hydrodynamic size (Dh) of P1 and P3 micelles was similar however P2 micelles were much smaller (Table 1). Size distribution curves based on intensity average for all polymers were bimodal, however in case of P2 the peak at lower size (10–40 nm) was the major component while the peak at larger size (70–300 nm) was major for P1 (Fig. 2b and d) even though both P1 and P2 have same position and number of azobenzene units. The smaller size of P2 micelles among all polymers is probably due to higher hydrophobicity of dodecyl chains and compact nature of the dendron while the largest size for P4 was probably due to highly hydrophobic G3 dendron. | ||
| Fig. 2 TEM images (a and c) and DLS size distribution curves (b and d) for 0.1 wt% aqueous solution of P1 and P2, respectively. | ||
| Polymer | Wphilic | Dh (nm) | PDI | cmc (M) × 10−5 |
|---|---|---|---|---|
| P1 | 62.8 | 77 | 0.30 | 2.50 |
| P2 | 57.3 | 26 | 0.41 | 1.25 |
| P3 | 51.5 | 70 | 0.54 | 0.89 |
| P4 | 39.5 | 139 | 0.10 | 1.25 |
Critical micelle concentration (cmc) of polymer assemblies was determined by using Nile red as hydrophobic solvatochromic probe because its absorption and emission maxima are well separated from azobenzene absorption spectra. Nile red exhibits weak fluorescence emission in water, however when in nonpolar environment such as the micellar core, its emission intensity increases significantly. Hence, cmc can be determined from a plot of fluorescence intensity versus log(polymer conc.) (Fig. S37†). The values for cmc are in the range of 10−5 M that are typical for amphiphilic macromolecules (Table 1).
Light-responsive behaviour
Light-responsive behaviour of the aggregates was investigated by irradiating the aqueous solution (0.02 wt%) with UV and visible light. For trans-to-cis photoisomerisation of azobenzene the solution was irradiated with 365 nm light (8 W lamp) and UV-vis spectra were recorded at different irradiation times.For P1, P2 and P4 absorption at the π–π* transition at 350 nm decreased with time and the weak n–π* transition at 450 nm increased indicating the isomerisation of azobenzene from trans to cis form (Fig. S38†). UV-vis spectra of P3 revealed presence of H-type aggregates of azobenzene in the micellar core by the presence of characteristic blue-shifted π–π* band with λmax at 308 nm (Fig. 3).35 A shoulder at 350 nm suggested the presence of free azobenzene that is, azobenzene units that were not part of H-type aggregates. Co-existence of H-aggregates and non-aggregated azobenzenes in small molecule as well as polymer-based assemblies is well documented.26,36,37 Irradiation of P3 solution at 365 nm resulted in decrease of absorbance of 308 nm and 350 nm peaks and increase in the absorbance at 450 nm peak indicating trans–cis isomerisation of free as well as bound (H-aggregate) azobenzene chromophores. The extent of trans to cis isomerisation (photostationary state, PSS) of azobenzene was calculated using Fisher method38 to be 73%, 79%, 55% and 65% for P1, P2, P3 and P4, respectively (Table 2). The higher value of PSS for P2 may be due to smaller size of the aggregate that allows greater ability to isomerise due to smaller aggregation number.39 Next, we investigated the reverse process, that is, cis-to-trans isomerisation of azobenzene by irradiating with visible light (450 nm, 6 W).40–42 The absorbance of π–π* band increased and that of n–π* transition peak decreased with irradiation time, which indicated transition of azobenzene from cis to trans form. The photoisomerisation ratio (% PSS) was observed to follow the same trend as for trans-to-cis isomerisation. Thus, it was demonstrated that the assemblies are photoresponsive and azobenzene unit in the hydrophobic core does undergo photoisomerisation although the process is hindered due to the aggregation in micellar core with more constraints in case of G3 dendron. Azobenzene units in H-aggregate also underwent photoisomerisation without disrupting the aggregate.41 The assemblies were stable after photoirradiation as suggested by size analysis using DLS (Fig. S39†).
 | ||
| Fig. 3 UV-vis spectra of 0.02 wt% aqueous solution of P3 for irradiation with (a) 365 nm (8 W lamp) and (b) 450 nm (6 W lamp) light. | ||
| Polymer | % photo-isomerisation (trans–cis) | % photo-isomerisation (cis–trans) | % thermal isomerisation (cis–trans) | k (s−1) × 10−5 |
|---|---|---|---|---|
| P1 | 73 | 70 | 62 | 1.69 |
| P2 | 79 | 79 | 63 | 1.84 |
| P3 | 55 | 50 | 26 | 1.59 |
| P4 | 65 | 60 | 52 | 3.05 |
H-Type aggregates of azobenzene present in the micellar assemblies of P3 may form during micellisation as the hydrophobic dendrons associate upon addition of water to THF. To understand this process, UV-vis spectra were recorded during the preparation of aqueous solution of P3 from THF via dialysis.
The λmax was at 350 nm in THF solution showing complete absence of H-type aggregates. Upon addition of water to solution of P3 in THF the peak at 350 nm shifted with broadening to 325 nm at THF:water 1:3 (v/v) composition (Fig. 4). Concentration of the polymer was same as that used for all photoisomerisation experiments (0.02 wt%). This solution was then dialysed against deionized water and UV-vis spectra were recorded with time. The peak at 325 nm further shifted to 308 nm after only 1 h of dialysis. Continuation of dialysis resulted in increase in absorbance of the peak that reached saturation after 25 h. In a control experiment, UV-vis spectra of P2 in THF:water (1:3 v/v) were recorded and no shift in the peak at 350 nm was observed (Fig. S40†). Thus, it was ascertained that the formation of H-aggregates takes place for P3 during micellisation when water content is higher than 75%.
 | ||
| Fig. 4 UV-vis spectra of 0.02 wt% solution of P3 in THF/water during dialysis against water. | ||
Thermal cis–trans isomerisation process of azobenzene is much slower than under photoirradiation and hence allows the study of kinetics of this process.43,44 To understand the effect of dendron structure on the kinetics of photoisomerisation, first-order rate constants were calculated for thermal cis–trans isomerisation. Absorption spectra were recorded at different time intervals during irradiation of polymer solution at 365 nm as well as during the storage in dark for 24 h after reaching photostationary state (Fig. S41†). The rate constant was calculated by fitting the experimental data to the following equation34
| ln(A∞ − At)/(A∞ − A0) = −kt |
First order rate constants for thermal isomerisation of all polymers are given in Table 2. Also, the photoisomerisation rates as expressed in terms of rate constants are comparable with those seen in azobenzene containing amphiphilic molecules where the movement of azobenzenes in the micellar core is limited.18,45
Dye encapsulation and light-responsive controlled release
Encapsulation and light-responsive controlled release of a hydrophobic dye from the polymer micelles was investigated as a function of dendron structure. Extent of encapsulation of the dye in polymer micelles was determined from UV-vis spectra of Nile red following the solid phase extraction method (Fig. S43†).46 For calculations, the reported value of molar extinction coefficient of Nile red in methanol (45000 M−1 cm−1)47 was used and the values for encapsulation capacity (defined as mmol of dye per mol of polymer) are plotted in Fig. 5. Encapsulation ability of the assemblies can also be expressed as encapsulation efficiency (mg of dye per g of polymer), plot for which is given in ESI (Fig. S44†). The values for P1, P3 and P4 were higher than for P2 probably due to higher number of aromatic rings that can better accommodate the aromatic heterocyclic dye through π–π stacking (Table S1†).
 | ||
| Fig. 5 Plot for encapsulation capacity of 0.5 wt% aqueous solution of P1–P4 using Nile red. | ||
Before studying the fate of encapsulated Nile red upon irradiation with UV and visible light, it was important to confirm the absence of its photobleaching. A solution of Nile red in methanol:water (1:1) was irradiated with 365 nm and 450 nm light separately, and fluorescence spectra were recorded at different time intervals. Fluorescence emission spectra of Nile red were found to change negligibly over 30 minutes of irradiation suggesting that any change in fluorescence emission intensity observed during dye release experiments involving multiple photoisomerisation cycles will be due to change in environment of the dye and not due to photobleaching (Fig. S45†).
When azobenzene is isomerised to cis form, the environment of micellar core turns polar and the encapsulated hydrophobic dye is released. When it reverts back to trans form the micellar environment regains hydrophobicity and some of the dye may re-enter the core affecting the net release, provided the micellar assembly is intact after trans–cis isomerisation. cis–trans isomerisation can be carried out in two different ways and although the percentage of PSS is similar in both the pathways the rates are very different. It is worthwhile to study whether the pathway followed for cis–trans isomerisation influences the net dye release. To investigate this possibility, Nile red loaded polymer solution was irradiated with 365 nm UV light for 30 minutes and fluorescence emission spectra of Nile red were recorded by exciting at 550 nm. It was observed that emission intensity at 630 nm decreased for all polymer solutions indicating that the polarity of environment of the dye increased. This decrease in intensity could be due to either disruption of the micelle and release of the dye from the micellar core into the aqueous solution or change in the polarity of the core due to presence of more polar cis isomer of azobenzene without release of the dye.48 cis–trans isomerisation was then carried out by irradiation with visible light as well as by storing the sample in dark and fluorescence spectra of Nile red were monitored. Emission intensity did not reach the initial value in both pathways suggesting leakage of dye from micellar core. Comparison of decrease in emission intensity of Nile red following two pathways for cis–trans isomerisation is shown in Fig. 6a. Since dye release cannot be directly quantified from fluorescence spectra due to several factors such as entrapment of dye in the PEG corona, the decrease in intensity may be considered as an approximate measure of the dye release.49
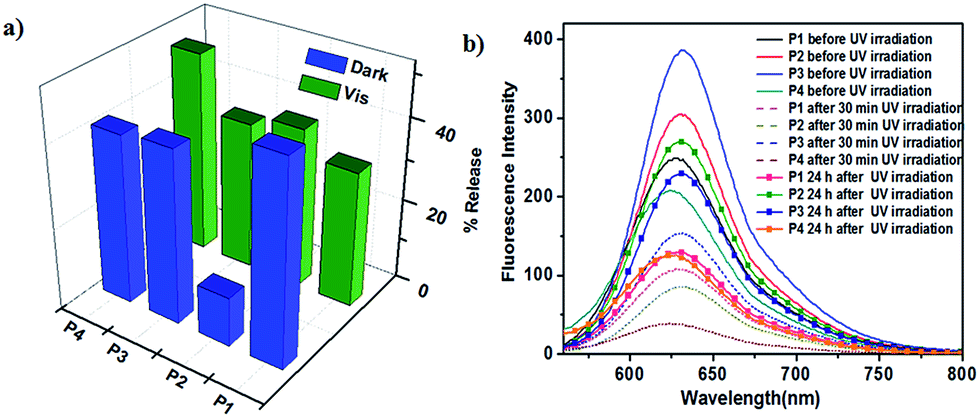 | ||
| Fig. 6 (a) Nile red release from aqueous solution of polymers following two different pathways for cis–trans photoisomerisation (b) emission spectra of Nile red at different intervals of time during UV-thermal photoisomerisation. | ||
Decrease in intensity for thermal isomerisation pathway was found to be higher than after photoisomerisation using visible light for P1 and P3, however the trend was reversed in case of P2 and P4 (Table S2†). In fact, decrease in Nile red intensity for thermal isomerisation for P2 was significantly lower, only 12%. Dye release for P4 was the highest in photoisomerisation pathway. This may be attributed to the difference in the dendron structure that is, different peripheral groups and number of azobenzene units, which in turn directs the size and nature of micellar assembly. Fluorescence emission spectra of Nile red for the photo-thermal trans–cis–trans isomerisation pathway are shown in Fig. 6b. Thus, it seems feasible to exert control over dye release by selecting desired pathway for cis–trans isomerisation of azobenzene.
To explore another approach for controlled release of dye, multiple photoisomerisation cycles were performed with thermal restoration gap between two cycles.18 A trans–cis–trans isomerisation carried out by irradiation of 365 nm light followed by 450 nm light is considered as one photocycle and the time allowed for dye to equilibrate between micellar core and outer aqueous environment when the sample is stored in dark is called thermal restoration period.
After the first photocycle there was a net decrease in Nile red intensity for all polymers. However, after second consecutive photocycle, that is, without any thermal restoration, no net decrease in intensity was observed except for P4, which showed slight increase. The sample was then kept for thermal restoration for 12 hours after which one photocycle was performed. Interestingly, some of the dye had re-entered the micellar core during thermal restoration period as seen from emission spectra at the beginning of photocycle. After the photocycle net decrease in intensity was less than after the first two photocycles. Thus, there was a net re-encapsulation of Nile red by micellar assemblies and the effect was more pronounced for P3. Polymer P4 however did not show any further release or re-encapsulation after thermal restoration. After another 4 hours of thermal restoration followed by the fourth photocycle there was a net decrease in intensity that was only slightly higher than that after the first photocycle with value for P4 remaining the same. Overall, after four photocycles and two thermal restoration periods only about 35–40% decrease in emission intensity of the dye was observed for P1–P3 while P4 showed ∼50% decrease in intensity and most of the dye release occurred after the first photocycle only (Fig. 7). In a similar experiment, ∼80% Nile red release was observed from assemblies of a linear polyester with PEG in the main chain and azobenzene in the side chain.18 Controlled photoinduced dye release from membranes of vesicles assembled from azobenzene-containing linear-dendritic copolymers has also been shown but not quantified.26 Thus, although the linear-dendritic copolymer micelles studied here show a strong tendency to retain the dye in general, there is significant difference in dye release profiles owing to difference in dendron structure. Fluorescence emission spectra for all photocycles are given in ESI (Fig. S46–S49†).
 | ||
| Fig. 7 Release of Nile red for polymers P1–P4 during different photocycles and with different thermal restoration intervals (NTR: no thermal restoration, TR: thermal restoration). | ||
Conclusions
Linear-dendritic copolymers with azobenzene in the interior and different hydrophobic groups at the periphery of second and third generation dendrons were synthesised and self-assembled in aqueous solution. Polymer containing azobenzenes on dendron periphery showed H-type aggregation in micelles that remained stable during photoisomerisation. H-Aggregates formed during micellisation as confirmed by following the dialysis of THF:water mixture using UV-vis spectroscopy. Polymer micelles were found to be highly stable during the photoisomerisation. Rate constants for thermal cis–trans isomerisation were comparable with those in azobenzene-containing amphiphilic polymers. Release of Nile red due to change in polarity of micellar core as indicated by decrease in fluorescence emission intensity clearly depended on the pathway used. This is the first time that a comparison of different isomerisation pathways for dye release from amphiphilic linear-dendritic copolymer assemblies has been made. Dye release studied over multiple photocycles interspersed with thermal restoration period also revealed good dye retention ability of the linear-dendritic copolymers. P4 aggregates showed higher release than other polymer micelles possibly due to higher generation dendron whereas P2 micelles displayed much lower release during a UV-thermal isomerisation cycle compared to the other polymers. Both the dye release experiments, clearly demonstrated the effect of dendron structure with significant differences seen in dye release profiles of the polymer assemblies and hence show fine control over dye release. Such structural tuning in photoresponsive polymers may contribute towards the development of better controlled release systems.
Acknowledgements
NK thanks UGC for research fellowship. AVA acknowledges financial support from SERB, Department of Science and Technology, New Delhi under the Fast Track Scheme (SR/FT/CS-79/2010).Notes and references
- S. Bai and Y. Zhao, Macromolecules, 2001, 34, 9032 CrossRef CAS.
- S. Sévigny, L. Bouchard, S. Motallebi and Y. Zhao, Macromolecules, 2003, 36, 9033 CrossRef.
- Y. Zhao and X. Tong, Adv. Mater., 2003, 15, 1431 CrossRef CAS.
- Y. Wu, A. Kanazawa, T. Shiono, T. Ikeda and Q. Zhang, Polymer, 1999, 40, 4787 CrossRef CAS.
- T. Breiner, K. Kreger, R. Hagen, M. Häckel, L. Kador, A. H. E. Müller, E. J. Krammer and H. W. Schmidt, Macromolecules, 2007, 40, 2100 CrossRef CAS.
- E. Blasco, B. V. K. J. Schmidt, C. Barner-Kowollik, M. Pinol and L. Oriol, Polym. Chem., 2013, 4, 4506 RSC.
- J. L. Mynar, A. P. Goodwin, J. A. Cohen, Y. Ma, G. R. Fleming and J. M. J. Fréchet, Chem. Commun., 2007, 2081 RSC.
- C. Zhu, C. Ninh and C. J. Bettinger, Biomacromolecules, 2014, 15, 3474 CrossRef CAS PubMed.
- G. Wang, X. Tong and Y. Zhao, Macromolecules, 2004, 37, 8911 CrossRef CAS.
- Z. Chen, Y. He, Y. Yang and X. Wang, Macromol. Rapid Commun., 2011, 32, 977 CrossRef CAS PubMed.
- H. Lee, W. Wu, J. Oh, L. Mueller, G. Sherwood, L. Peteanu, T. Kowalewski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2007, 46, 2453 CrossRef CAS PubMed.
- J. M. Schumers, C. A. Fustin and J.-F. Gohy, Macromol. Rapid Commun., 2010, 31, 1588 CrossRef CAS PubMed.
- J.-F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117 RSC.
- D. Wang and X. Wang, Prog. Polym. Sci., 2013, 38, 271 CrossRef CAS.
- L. Lin, Z. Yan, J. Gu, Y. Zhang, Z. Feng and Y. Yu, Macromol. Rapid Commun., 2009, 30, 1089 CrossRef CAS PubMed.
- E. Mabrouk, D. Cuvelier, F. Brochard-Wyart, P. Nassoy and M.-H. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7294 CrossRef CAS PubMed.
- X. Wang, Y. Yang, P. Gao, F. Yang, H. Shen, H. Guo and D. Wu, ACS Macro Lett., 2015, 4, 1321 CrossRef CAS.
- M. Kumari, M. Billamboz, E. Leonard, C. Len, C. Böttcher, A. K. Prasad, R. Haag and S. K. Sharma, RSC Adv., 2015, 5, 48301 RSC.
- R. Deloncle and A.-M. Caminade, J. Photochem. Photobiol., C, 2010, 11, 25 CrossRef CAS.
- B. Yu, X. Jiang, R. Wang and J. Yin, Macromolecules, 2010, 43, 10457 CrossRef CAS.
- M. Jin, R. Lu, C. Bao, T. Xu and Y. Zhao, Polymer, 2004, 45, 1125 CrossRef CAS.
- F. Wurm and H. Frey, Prog. Polym. Sci., 2011, 36, 1 CrossRef CAS.
- G. Whitton and E. R. Gillies, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5295 CrossRef.
- E. Blasco, M. Pinol and L. Oriol, Macromol. Rapid Commun., 2014, 35, 1090 CrossRef CAS PubMed.
- J. del Barrio, L. Oriol, C. Sánchez, J. L. Serrano, A. D. Cicco, P. Keller and M. H. Li, J. Am. Chem. Soc., 2010, 132, 3762 CrossRef CAS PubMed.
- E. Blasco, J. del Barrio, C. Sánchez-Somolinos, M. Piñol and L. Oriol, Polym. Chem., 2013, 4, 2246 RSC.
- E. Blasco, J. L. Serrano, M. Piñol and L. Oriol, Macromolecules, 2013, 46, 5951 CrossRef CAS.
- D. M. Junge and D. V. McGrath, J. Am. Chem. Soc., 1999, 121, 4912 CrossRef CAS.
- L.-X. Xiao, D. M. Junge and D. V. McGrath, Macromolecules, 2002, 35, 319 CrossRef.
- C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338 RSC.
- Z. Shi, H. Lu, Z. Chen, R. Cheng and D. Chen, Polymer, 2012, 53, 359 CrossRef CAS.
- Y. Yu, L. Zhang and A. Eisenberg, Macromolecules, 1998, 31, 1144 CrossRef CAS.
- Z. Feng, L. Lin, Z. Yan and Y. Yu, Macromol. Rapid Commun., 2010, 31, 640 CrossRef CAS PubMed.
- M. Shimomura and T. Kunitake, J. Am. Chem. Soc., 1987, 109, 5175 CrossRef CAS.
- W. Su, K. Han, Y. Luo, Z. Wang, Y. Li and Q. Zhang, Macromol. Chem. Phys., 2007, 208, 955 CrossRef CAS.
- E. Fisher, J. Phys. Chem., 1967, 71, 3704 CrossRef.
- C. Cördel, C. S. Popeney and R. Haag, Chem. Commun., 2011, 47, 6584 RSC.
- Z. Feng, L. Lin, Z. Yan and Y. Yu, Macromol. Rapid Commun., 2010, 31, 640 CrossRef CAS PubMed.
- G. Wang, X. Tong and Y. Zhao, Macromolecules, 2004, 37, 8911 CrossRef CAS.
- S. Wang, Q. Shen, M. H. Nawaz and W. Zhang, Polym. Chem., 2013, 4, 2151 RSC.
- P. Sierocki, H. Maas, P. Dragut, G. Richardt, F. Vögtle, L. D. Cola, F. Brouwer and J. I. Zink, J. Phys. Chem. B, 2006, 110, 24390 CrossRef CAS PubMed.
- J. Nithyanandhan, N. Jayaraman, R. Davis and S. Das, Chem.–Eur. J., 2004, 10, 689 CrossRef CAS PubMed.
- G. Wang, D. Yuan, T. Yuan, J. Dong, N. Feng and G. Han, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2768 CrossRef CAS.
- M. Wyszogrodzka and R. Haag, Chem.–Eur. J., 2008, 14, 9202 CrossRef CAS PubMed.
- M. Adeli, H. Nazami, F. Du, S. Hönzke, S. Headtrich, J. Keilitz and R. Haag, RSC Adv., 2015, 5, 14958 RSC.
- E. Blasco, B. V. K. J. Schmidt, C. Barner-Kowollik, M. Pinol and L. Oriol, Macromolecules, 2014, 47, 3693 CrossRef CAS.
- J. Jiang, X. Tong, D. Morris and Y. Zhao, Macromolecules, 2006, 39, 4633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterisation data and NMR, MALDI-TOF, UV-vis and fluorescence spectra. See DOI: 10.1039/c6ra02250b |
| This journal is © The Royal Society of Chemistry 2016 |