
Ternary cooperative Au–CdS–rGO hetero-nanostructures: synthesis with multi-interface control and their photoelectrochemical sensor applications†
Abstract
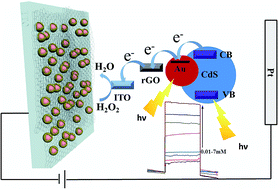
This paper demonstrates the synthesis of ternary cooperative semiconductor–metal–graphene (Au–CdS–rGO) hetero-nanostructures. The Au–CdS core–shell nanoparticles (∼50 nm) were sandwiched between reduced graphene oxide nanosheets. Decorating GO with L-cysteine hydrochloride plays an important role in sustaining the high level of dispersion of the Au–CdS nanoparticles (NPs). The as-fabricated photoanode exhibited higher photocurrent density compared to single-/binary-components, which could be attributed to the synergistic effect of ternary cooperative Au–CdS–rGO hetero-nanostructures. First, the hetero-nanostructure could facilitate more efficient utilization of incident light, confirmed by high incident-photon-to-current-conversion efficiency (IPCE). Then the photogenerated electrons could be efficiently transferred from CdS onto Au and graphene sheets, which benefits from the LSPR-induced charge separation at the interface of Au and CdS, and the electron-accepting ability of graphene. The obtained Au–CdS–rGO photoanode was used to quantify H2O2 concentration, and the detection limit was 0.005 mmol L−1, demonstrating promise for application in photoelectrochemical sensors.
 Please wait while we load your content...
Please wait while we load your content...

