
One-pot synthesis of acyloxy carbonyl compounds from ketones using a Pybox–copper(II) catalyst†
Wei-Guo Jia*,
Hui Zhang,
Dan-Dan Li and
Li-Qin Yan*
College of Chemistry and Materials Science, Center for Nano Science and Technology, The Key Laboratory of Functional Molecular Solids, Ministry of Education, Anhui Laboratory of Molecular-Based Materials, Anhui Normal University, Wuhu, 241000, China. E-mail: wgjiasy@mail.ahnu.edu.cn; yanliqin@mail.ahnu.edu.cn
First published on 1st March 2016
Abstract
We have described the first efficient one-pot method for the synthesis of acyloxy carbonyl compounds from ketones catalyzed by a Pybox–Cu(II) complex under mild conditions. A series of α-acyloxy ketone products were obtained in good to excellent yields.
Acyloxy ketones are important organic synthetic intermediates in organic synthesis and versatile building blocks for natural products and pharmaceuticals.1 Thus, many efforts have been devoted to finding efficient synthetic approaches to α-acyloxy ketones and their derivatives using different synthetic methods by a large number of research groups. A variety of protocols have been reported including transition metal-catalyzed and organocatalytic methods. In 1998, Ohfune and co-workers reported the copper-catalyzed insertion of α-diazoketones into carboxylic acids to give α-acyloxy ketones.2 In 2009, the Cadierno group described the addition of carboxylic acids to propargyl alcohols leading to the formation of α-acyloxy ketones, which was catalyzed by Ru complexes.3 However, in contrast to transition metal catalysts, organocatalysts have been more extensively explored for the catalyzing the α-acyloxylation of ketones, with examples including the dimsyl anion,4 (hypo)iodite,5 N-heterocyclic carbenes (NHCs),6 amines,7 2-tritylpyrrolidine8 as well as Bu4NI.9 Although these organocatalyst-based reactions are robust and useful for the synthesis of a variety of α-acyloxyl carbonyl compound architectures containing a range of functional groups, the transition metal-catalyzed reaction of propiophenone to generate α-acyloxylation products has not been reported.
Herein, we have reported an efficient one-pot method for the synthesis of α-acyloxy carbonyl compounds in the presence of both moisture and air, catalyzed by a Pybox–Cu(II) complex. Treatment of a variety of ketones with [(Dm-Pybox)Cu(II)Br2] and K4[Fe(CN)6]·3H2O provides the α-acyloxy ketone products in isolated yield of 67–97%. The transformation is regiospecific in the discrimination of electron-deficient intermediate, in the case of non-symmetrical substrates.
Recently, we developed an efficient method for the synthesis of α-amino ketones and esters under aerobic oxidative conditions using a [(Dm-Pybox)CuBr2] complex as a catalyst, which proceeded through an α-bromo carbonyl intermediate.10 In our continued effort to develop environmentally benign protocols for the synthesis of biologically and pharmacologically active compounds,11 we attempted to extend this nucleophile-catalyzed aerobic oxidation protocol to the synthesis of aromatic α-cyano ketones from ketones with potassium hexacyanoferrate as a CN− source.12 In this previous work, when propiophenone and potassium hexacyanoferrate were combined under the reaction conditions the expected compound, 2-methyl-3-oxo-3-phenylpropanenitrile, was not observed. Instead, an unexpected compound was obtained as the major product.
With this rather unexpected result in hand, we started our study using the model reaction shown in Table 1: propiophenone (0.5 mmol) and potassium hexacyanoferrate(II) (2.4 equiv.) in DMF (1 mL). However, no reaction took place after heating the mixture at 110 °C for 9 h either with or without any ligand (Table 1, entry 1 and entry 2, L = Dm-Pybox). The α-acyloxylation product was obtained in only 48% yield when using CuBr2 as a catalyst (Table 1, entry 3), however, to our delight, 90% yields of the desired product were observed when using 2,6-bis[4′,4′-dimethyloxazolin-2′-yl]pyridine (Dm-Pybox) as the ligand (Table 1, entry 4). We then chose [(Dm-Pybox)Cu(II)Br2] complex as the catalyst to test the influence of solvents on the reaction yield (Table 1, entries 5–7). To our delight, DMF was the best solvent for this transformation. However, a lower yield was obtained with a lower catalyst loading of 5 mol% [(Dm-Pybox)Cu(II)Br2] and it required a longer reaction time of 16 hours (Table 1, entry 8). The yield remained unchanged when using 15 mol% [(Dm-Pybox)Cu(II)Br2], but the reaction time was shortened to 6 hours (Table 1, entry 9). In order to ascertain that O2 was necessary (as shown in proposed mechanism later in Scheme 2) for the reaction to proceed, a control reaction was carried out in presence of N2. In the absence of O2, only 9% of the desired product was obtained (Table 1, entry 10), which clearly demonstrated the role that O2 played in this reaction. Finally, the amount of K4[Fe(CN)6]·3H2O was also surveyed, with only 9% of the desired product obtained in the presence of 10 mol% CN− source (Table 1, entry 11). It is proven that an equivalent of K4[Fe(CN)6]·3H2O was necessary for the reaction to proceed. A control experiment with 20 mol% TEMPO showed no reaction (Table 1, entry 12). This result might support the formation of an intermediate, E, in the radical mechanism pathway shown in Scheme 2.
|
||||
|---|---|---|---|---|
| Entry | Cu catalyst (10 mol%) | Ligand (mol%) | Solvent | Yielda,b (%) |
| a Isolated yield.b Reaction conditions: 1 mL DMF as solvent, under air, 110 °C.c 5 mol% catalyst, reaction time: 16 h.d 15 mol% catalyst, reaction time: 6 h.e Under N2.f 0.0083 mmol K4[Fe(CN)6]·3H2O.g 20 mol% TEMPO was added, NR = no reaction.h 1.2 mmol CuCN.i 0.6 mmol Zn(CN)2. | ||||
| 1 | DMF | NR | ||
| 2 | L | DMF | NR | |
| 3 | CuBr2 | DMF | 48 | |
| 4 | Cu complex | DMF | 90 | |
| 5 | Cu complex | DMSO | 11 | |
| 6 | Cu complex | NMP | NR | |
| 7 | Cu complex | DMF | 91 | |
| 8 | Cu complex | DMF | 73c | |
| 9 | Cu complex | DMF | 92d | |
| 10 | Cu complex | DMF | 9e | |
| 11 | Cu complex | DMF | 9f | |
| 12 | Cu complex | DMF | NRg | |
| 12 | Cu complex | DMF | 50h | |
| 12 | Cu complex | DMF | 33i | |

 | ||
| Scheme 1 The gram-scale synthesis of 2a. | ||
 | ||
| Scheme 2 A proposed catalytic mechanism for the α-acyloxylation of ketones. | ||
In order to explore the generality and scope of the present α-acyloxylation method, several ketone compounds were examined as substrates using the optimized reaction conditions. Propiophenone derivatives, which were substituted with electron-donating or electron-withdrawing groups, gave the corresponding α-benzoyloxy ketones 1a–5a in good to excellent yields. The configuration of 1a was determined by X-ray crystallographic analysis (Fig. 1). Interestingly, the efficient conversion of electron-deficient ketones was achieved at low temperature (Table 2, entry 5). Notably, 2-butyrylfuran and 2-butyrylthiophene were also tested, and these reactions proceeded smoothly to give 6a and 7a in a good and moderate yield, respectively, in the presence of 15 mol% of the [(Dm-Pybox)Cu(II)Br2] catalyst.
 | ||
| Fig. 1 The molecular structure of 1a with thermal ellipsoids drawn at the 30% level. All hydrogen atoms are omitted for clarity. | ||













In general, the selectivity of the reaction is determined by electronic effects, so control experiments were carried out to test the selectivity of α-acyloxylation reaction. Equimolar amounts of 1-p-tolylpropan-1-one and 1-(4-chlorophenyl)propan-1-one were heated in the presence of 10 mol% [(Dm-Pybox)Cu(II)Br2] catalyst and 2.4 equivalents of CN− in DMF to give 8a in 88% yield (Table 3). No other side products were observed in this transformation. The configuration of 8a was also determined by X-ray crystallographic analysis (Fig. 2). The electron-withdrawing group on the aromatic ring of 1-(4-chlorophenyl)propan-1-one more favors the formation of an intermediate that then results in the asymmetrical α-acyloxylation product. Propiophenone and 1-phenylbutan-1-one also transformed well under the standard conditions, and the desired products 1-oxo-1-phenylpropan-2-yl-4-chlorobenzoate (9a) and 1-oxo-1-phenylbutan-2-yl-4-chlorobenzoate (10a) were obtained in 71% and 89% yields, respectively (Table 3). Interestingly, the heterocyclic substrate 1-(thiophen-2-yl)butan-1-one underwent this transformation to provide 12a in acceptable yields.


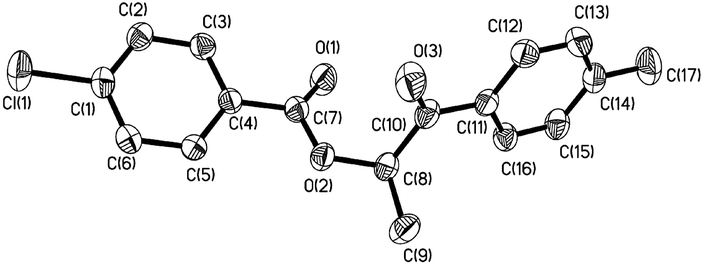 | ||
| Fig. 2 The molecular structure of 8a with thermal ellipsoids drawn at the 30% level. All hydrogen atoms are omitted for clarity. | ||
To further evaluate the practical utility of the catalyst system, a model reaction was carried out on a gram scale using the optimized conditions, and the desired product was obtained in 94% yield (Scheme 1).
To gain further insight into the reaction mechanisms of the Cu-catalyzed cross-coupling reaction, a different starting material, either benzoic acid or benzoyl cyanide, was added to investigate the viability of the intermediate. The desired products were observed in both cases indicating the involvement of benzoic acid and benzoyl cyanide in the mechanistic process (see ESI†). Based on documented precedent and experiment results, a reaction mechanism was proposed as shown in Scheme 2. Initially, a cyanohydrin intermediate A is formed by cyanide addition to propiophenone. Intermediate A can then undergo oxidation to yield benzoyl cyanide B under aerobic oxidation conditions, which is hydrolyzed in situ to give the benzoic acid (C) in wet organic solvents,13 then decarbonylation results in the formation of (D). Meanwhile, propiophenone can undergo bromination at the α-carbonyl to generate 2-bromo-1-phenylpropan-1-one (E) in the presence of the [(Dm-Pybox)Cu(II)Br2] catalyst.14 Intermediate E is then further hydrolyzed to afford 2-hydroxy-1-phenylpropan-1-one (F), which can react with intermediate D to afford the final product. The existence of intermediate F is supported by the MS spectra. In addition, the Cu(II) catalyst is regenerated by the oxygen-mediated reoxidation of Cu(I).
Conclusions
In conclusion, we have described the first efficient and one-pot green method for the synthesis of α-acyloxy carbonyl compounds from ketones catalyzed by a Pybox–Cu(II) complex under mild conditions. A series of α-acyloxylation products were obtained in good to excellent yields. This method provides an excellent alternative to organocatalysts. A detailed reaction mechanism is still under investigation.Acknowledgements
This work was supported by the National Nature Science Foundation of China (21102004), the NSF of Anhui province (1208085MB25) and the Project-sponsored by SRF for ROCS and Special and Excellent Research Fund of Anhui Normal University. We are grateful to Prof. Shaowu Wang and Zhuo Chai for their valuable suggestions.Notes and references
- (a) M. Shindo, Y. Yoshimura, M. Hayashi, H. Soejima, T. Yoshikawa, K. Matsumoto and K. Shishido, Org. Lett., 2007, 9, 1963–1966 CrossRef CAS PubMed; (b) W. W. Pei, S. H. Li, X. P. Nie, Y. W. Li, J. Pei, B. Z. Chen, J. Wu and X. L. Ye, Synthesis, 1998, 1298–1304 CrossRef CAS; (c) C. M. Loner, F. A. Luzzio and D. R. Demuth, Tetrahedron Lett., 2012, 53, 5641–5644 CrossRef CAS PubMed.
- T. Shinada, T. Kawakami, H. Sakai, I. Takada and Y. Ohfune, Tetrahedron Lett., 1998, 39, 3757–3760 CrossRef CAS.
- (a) V. Cadierno, J. Francos and J. Gimeno, Green Chem., 2010, 12, 135–143 RSC; (b) V. Cadierno, J. Francos and J. Gimeno, Organometallics, 2011, 30, 852–862 CrossRef CAS; (c) D. Devanne, C. Ruppin and P. H. Dixneuf, J. Org. Chem., 1988, 53, 925–926 CrossRef CAS.
- O. Bortolini, G. Fantin, V. Ferretti, M. Fogagnolo, P. P. Giovannini, A. Massi, S. Pacifico and D. Ragno, Adv. Synth. Catal., 2013, 355, 3244–3252 CrossRef CAS.
- M. Uyanik, D. Suzuki, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2011, 50, 5331–5334 CrossRef CAS PubMed.
- R. N. Reddi, P. V. Malekar and A. Sudalai, Org. Biomol. Chem., 2013, 11, 6477–6482 CAS.
- (a) C. S. Beshara, A. Hall, R. L. Jenkins, K. L. Jones, T. C. Jones, N. M. Killeen, P. H. Taylor, S. P. Thomas and N. C. O. Tomkinson, Org. Lett., 2005, 7, 5729–5732 CrossRef CAS PubMed; (b) D. A. Smithen, C. J. Mathews and N. C. O. Tomkinson, Org. Biomol. Chem., 2012, 10, 3756–3762 RSC.
- T. Kano, H. Mii and K. Maruoka, J. Am. Chem. Soc., 2009, 131, 3450–3451 CrossRef CAS PubMed.
- S. Guo, J.-T. Yu, Q. Dai, H. Yang and J. Cheng, Chem. Commun., 2014, 50, 6240–6242 RSC.
- W.-G. Jia, D.-D. Li, Y.-C. Dai, H. Zhang, L.-Q. Yan, E.-H. Sheng, Y. Wei, X.-L. Mu and K.-W. Huang, Org. Biomol. Chem., 2014, 12, 5509–5516 CAS.
- (a) A. Park and S. Lee, Org. Lett., 2012, 14, 1118–1121 CrossRef CAS PubMed; (b) J. Shen, D. Yang, Y. Liu, S. Qin, J. Zhang, J. Sun, C. Liu, C. Liu, X. Zhao, C. Chu and R. Liu, Org. Lett., 2014, 16, 350–353 CrossRef CAS PubMed.
- (a) T. Chatterjee, R. Dey and B. C. Ranu, J. Org. Chem., 2014, 79, 5875–5879 CrossRef CAS PubMed; (b) D. Zhang, H. Sun, L. Zhang, Y. Zhou, C. Li, H. Jiang, K. Chen and H. Liu, Chem. Commun., 2012, 48, 2909–2911 RSC; (c) T. D. Senecal, W. Shu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 10035–10039 CrossRef CAS PubMed.
- Y.-J. Kim, N. Y. Kim and C.-H. Cheon, Org. Lett., 2014, 16, 2514–2517 CrossRef CAS PubMed.
- R. W. Evans, J. R. Zbieg, S. Zhu, W. Li and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 16074–16077 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional copies of NMR spectra. Crystallographic information files (CIFs) CCDC 1012733 and 1012734 for complex 1a and 8a. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra02186g |
| This journal is © The Royal Society of Chemistry 2016 |