
Fabrication of poly(cyclotriphosphazene-co-4,4′-sulfonyldiphenol) nanotubes decorated with Ag–Au bimetallic nanoparticles with enhanced catalytic activity for the reduction of 4-nitrophenol
Ya Yan,
Jianwei Fu*,
Minghuan Wang,
Shujun Liu,
Qianqian Xin,
Zhimin Chen and
Qun Xu*
School of Materials Science and Engineering, Zhengzhou University, Zhengzhou, P. R. China. E-mail: jwfu@zzu.edu.cn; qunxu@zzu.edu.cn; Fax: +86-371-67767827; Tel: +86-371-67767827
First published on 1st March 2016
Abstract
Ag–Au bimetallic nanoparticles (NPs) are deposited on poly(cyclotriphosphazene-co-4,4′-sulfonyldiphenol) (PZS) nanotubes by a facile and effective co-reduction method, wherein the PZS nanotubes with abundant hydroxyl groups have been prepared via an in situ template approach. Upon varying the feeding amounts of the Ag and Au precursors, the bimetallic compositions of the PZS nanotubes can be readily tuned resulting in a series of bimetallic catalysts with different Ag to Au molar ratios, thus leading to the tunable catalytic properties. Characterization results show that the Ag–Au bimetallic nanoparticles with smaller size and good dispersibility are well anchored onto the surface of the PZS nanotubes. Furthermore, the reduction of 4-nitrophenol (4-NP) into 4-aminophenol (4-AP) by NaBH4 is applied as a model reaction to study the effect of different Ag-to-Au molar ratios on the catalytic capabilities of the resulting composites. It is found that the catalytic capability is remarkably enhanced when the Au content is increased. The maximum activity parameter value reaches 92.2 s−1 g−1, which is far higher than that of PZS nanotubes decorated with either Ag or Au nanoparticles alone.
Introduction
Noble metal nanostructured materials have generated great interest primarily due to their fascinating physicochemical properties and potential applications in the fields of biomedicine, catalysis, photonics, electronics, and so on.1–6 In the last decade or so, many methods have been developed for the controllable design and synthesis of noble monometallic nanostructures.7–11 In contrast, little progress has been made in preparing well-defined bimetallic nanostructures, mostly because of the complicated and difficult approach required for the synthesis.12–14 Generally speaking, the synthesis of bimetallic compounds can be carried out in the solid, gaseous, or solution state. In comparison with the conventionally used solid and gaseous state routes (e.g. metallurgical techniques and molecular beam techniques), however, the solution-phase synthetic method of bimetallic nanocrystals (including co-reduction, seeded growth, the galvanic replacement reaction, and noble-metal-induced reduction) is more feasible in operation and allows for the controllable and simple synthesis of bimetallic nanocrystals with specific structure (core/shell, heterostructure, intermetallics, or alloys).15,16 Among them, the co-reduction strategy in solution system is a remarkably simple and effective method in preparing high-quality bimetallic NPs with tunable size and superior catalytic properties.17,18In bimetallic systems, they are expected to display not only a combination of the properties associated with two distinct metals, but also new properties due to a synergy between the two metals.19 Because of such synergistic effects, bimetallic catalysts can show significantly greater catalytic activity compared with monometallic catalysts, even at lower concentrations. For heterogeneous catalysis,20 noble bimetallic catalysts (e.g. Pt–Au21) have been investigated as catalyst to confer an unprecedentedly improved catalytic capability. It has been reported that the specific activity of bimetallic NPs is strongly related to their size and distribution. Highly distributed bimetallic NPs with smaller size and narrow distribution are ideal for high catalytic activity owing to their large surface-to-volume ratio. However, the small bimetallic NPs very easily aggregate during the catalytic or electrocatalytic reactions due to their higher surface energy, which could remarkably reduce their initial catalytic activity and selectivity.22 Therefore, various supporting materials such as carbon spheres, carbon nanotubes, SiO2, and polymers have been developed to stabilize bimetallic NPs.23–26
Among them, polymers have been considered as a promising candidate to stabilize metal nanoparticles. So far, many substrates with spherical or other morphologies have been chosen as supports, but few polymer nanotubes have been reported. As for polymer nanotubes, a new class of one-dimensional nanomaterials with high specific surface area, they have attracted tremendous attentions in recent years owing to their potential applications in various fields including optoelectronic nanodevices, chemical sensors, drug carriers.27 Generally, the fabrication of polymer nanotubes can be divided into two fundamental methods: self assembly and use of nanostrutured templates in a defined dimension.28–33 However, both self-assembly and template techniques are multistep processes. Therefore, it is a challenge and a prerequisite to develop facile methods to synthesize polymer nanotubes with controlled dimensions under mild conditions. Polyphosphazenes are a versatile class of novel organic/inorganic hybrid materials with the unique polyphosphazene inorganic backbone (–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) and structural multiplicity of side groups, including organic, organometallic or inorganic units,34–36 which makes them suitable for immobilizing metal ions, leading to a good dispersion of metallic NPs or bimetallic NPs after reduction.37,38 Therefore, it is very interesting to employ polyphosphazene to construct polymer nanotubes and load bimetallic NPs with catalytic activity.
N–) and structural multiplicity of side groups, including organic, organometallic or inorganic units,34–36 which makes them suitable for immobilizing metal ions, leading to a good dispersion of metallic NPs or bimetallic NPs after reduction.37,38 Therefore, it is very interesting to employ polyphosphazene to construct polymer nanotubes and load bimetallic NPs with catalytic activity.
In this work, a highly cross-linked polyphosphazene-poly(cyclotriphosphazene-co-4,4′-sulfonyldiphenol) (PZS) nanotube with abundant hydroxyl groups was firstly synthesized through an in situ template method. Then, using as-synthesized PZS nanotubes as stabilizer, we have fabricated PZS@Ag–Au NPs catalysts via a facile and effective co-reduction method in solution. The heteroatoms and a large number of hydroxyl groups of the PZS nanotubes39 plays a key role in the deposite of ultrafine and well distributed alloy nanoparticals by offering numerous coordinated sites for metal ions. This point has been well-proved in our previous works.40,41 Because of such coordination interaction effects, Ag–Au alloy NPs with a very narrow size distribution (about 2.6 nm) were successfully deposited on the PZS nanotubes. By varying the feeding amounts of AgNO3 and HAuCl4, a fine control was obtained over the synthesis of the Ag–Au NPs formed on the PZS nanotubes. It was found that the as-prepared PZS@Ag–Au NPs composites showed an excellent catalytic property towards the reduction of 4-nitrophenol (4-NP). Moreover, taking the advantage of the synergetic effect between Au and Ag, the obtained catalyst exhibits superior catalytic capability compared with monometallic PZS@Ag NPs composites and PZS@Au NPs composites. In light of these unique characteristics, the PZS@Ag–Au NPs composites might own great potential for practical applications.
Experimental
Materials
Hexachlorocyclotriphosphazene (HCCP) was purchased from Aldrich Chemical Co. Ltd. and sublimated twice before use. 4,4′-Sulfonydiphenol (BPS), silver nitrate (AgNO3), chloroauric acid (HAuCl4) were purchased from Sinopharm Chemical Reagent Co. Ltd. Tetrahydrofuran (THF), sodium borohydride (NaBH4), 4-nitrophenol (4-NP), and triethylamine (TEA) were from Tianjin Chemical Reagent Co. Ltd.Synthesis of the PZS nanotubes
The facile preparation of PZS nanotubes was carried out as follows: 50 mL THF with HCCP (0.4 g, 1.152 mmol) was added dropwise into 50 mL THF with BPS (1.29 g, 5.16 mmol) and TEA (1.04 g, 10.328 mmol). The reaction mixtures were stirred in an ultrasonic bath (50 W, 80 kHz) at 40 °C for 12 h. The yielded solid was centrifuged and then washed three times with THF and deionized water respectively. Then the solid was dried under vacuum to yield PZS nanotubes as a white powder.Preparation of PZS@Ag–Au NPs composites
PZS@Ag–Au NPs composites were prepared by the co-reduction method. Typically, AgNO3 (20 mM) and HAuCl4 (20 mM) was added to an aqueous PZS nanotubes dispersion (20.0 mL, 0.5 mg mL−1). The mixture was stirred in an ultrasonic bath (190 W) at room temperature for 30 min. Then on a magnetic stirrer at room temperature, 0.01 wt% NaBH4 solution was added dropwise into flask in the presence of trisodium citrate within 5 min. As soon as the reaction was completed, the produced solid was washed several times with deionized water and ethanol, respectively. Finally, the collected product were dried under the nitrogen atmosphere to obtain the PZS@Ag–Au NPs composites.To carry out the comparative study, the feeding amount of AgNO3 (20 mM, 0.2 mL) was fixed in all experiments, while changing the amount of HAuCl4 (20 mM) to produce normalized Ag to Au ratios at 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 4:1, 2:1, 4:3 and 1:1. Monometallic catalysts were also prepared by the same method, except that a different amount of metal precursor was used (0.2 mL AgNO3 20 mM and 0.2 mL HAuCl4 20 mM).
:1, 4:1, 2:1, 4:3 and 1:1. Monometallic catalysts were also prepared by the same method, except that a different amount of metal precursor was used (0.2 mL AgNO3 20 mM and 0.2 mL HAuCl4 20 mM).
Catalytic reduction of 4-nitrophenol (4-NP)
Typically, an aqueous solution of NaBH4 (1.0 mL, 7.5 × 10−3 mol L−1) was mixed with aqueous 4-NP solution (1.7 mL, 1.0 × 10−4 mol L−1) in the quartz cell (1 cm path length), leading to a color change from light yellow to yellow-green. Then, PZS@Ag–Au NPs composites, PZS@Ag NPs composites or PZS@Au NPs composites (0.1 mL, 1 mg 2 mL−1) were added, and the mixture solution was quickly measured by UV-vis spectroscopy. The progress of the conversion of 4-NP to 4-AP was then monitored via UV-vis spectroscopy by recording the time-dependent absorbance spectra of the reaction mixture in a scanning range 200–600 nm at ambient temperature.Characterization
The morphologies of the PZS nanotubes, PZS@Ag NPs composites, PZS@Au NPs composites and PZS@Ag–Au NPs composites were examined by scanning electron microscopy (SEM, JSM 6700F, JEOL) and transmission electron microscopy with an accelerating voltage of 100 kV (TEM, Tecnai G2 20 S-TWIN, FEI). The X-ray diffraction (XRD) patterns were recorded on a Netherlands X'Pert PRO X-ray diffractometer. The products were recorded in the 2θ range from 10.0° to 90° in steps of 0.03 with a count time of 5 s each time. The X-ray photoelectron spectroscopy (XPS) measurement was carried out on a RBD upgraded PHI-5000C ESCA system (Perkin Elmer). Atomic absorption spectroscopy (AAS) was performed on Fuli AA170 spectrometer. UV-vis spectroscopy was performed on Shimadzu UV-240 spectrometer ranging from 200 to 600 nm.Results and discussion
Fig. 1 illustrates the fabrication of the PZS@Ag–Au NPs composites. Firstly, using HCCP and BPS as comonomers and TEA as acid-acceptor, cross-linked organic–inorganic hybrid PZS nanotubes with abundant hydroxyl groups were successfully synthesized according to a modified in situ-template approach.39 Then, using as-synthesized PZS nanotubes as a support, and AgNO3 and HAuCl4 as noble metal precursors, the PZS@Ag–Au NPs composites were prepared through the co-reduction approach. Finally, the catalytic activity of as-prepared bimetallic catalysts was investigated by employing the reduction of 4-nitrophenol (4-NP) into 4-aminophenol (4-AP) by NaBH4 as a model reaction.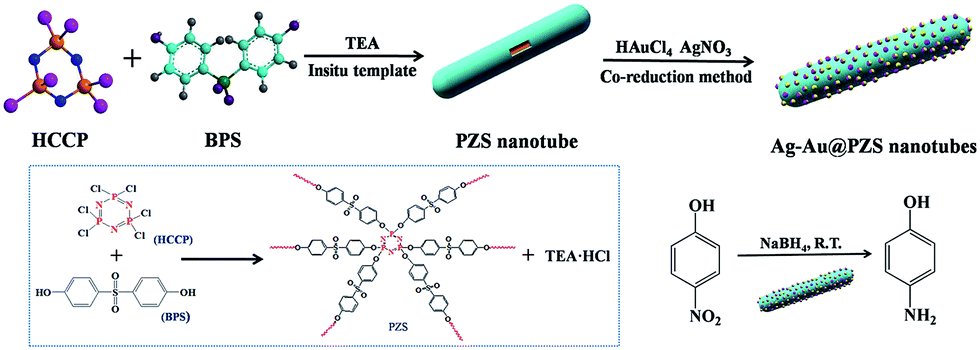 | ||
| Fig. 1 Fabrication scheme of the PZS@Ag–Au NPs composites and the reduced protocol of 4-nitrophenol by NaBH4 using PZS@Ag–Au NPs composites as a catalyst. | ||
Fig. 2a and b show the SEM images of as-synthesized PZS nanotubes with abundant hydroxyl groups. Obviously, the PZS nanotubes are short rods with uniform lengths of 2–4 μm, and most of them have an outer diameter in the range of 90–120 nm. The TEM image (Fig. 2c and d) reveals that the nanotubes possess hollow tubular structures with an inner diameter of about 20–30 nm and every tube's inner diameter are uniform. Meanwhile, it should be noted that most of the nanotube ends are closed, which might result from ultrasonic irradiation with higher power exerted on the reaction system.
 | ||
| Fig. 2 SEM and TEM images of the PZS nanotubes. | ||
Then, the PZS nanotubes, a class of functional polymer nanotubes, were used as supporters to in situ synthesize PZS@Ag NPs composites, PZS@Au NPs composites, and a series of PZS@Ag–Au NPs composites with sodium borohydride as the reducing reagent. Fig. 3a and b show the representative TEM images of the as-synthesized PZS@Ag NPs composites and PZS@Au NPs composites, respectively. It is obvious that metal NPs are uniformly dispersed on the outer walls of the PZS nanotubes. Additionally, according to the particle size distribution histogram, the average size of Ag NPs and Au NPs are 2.8 nm and 3.6 nm respectively, suggesting the monodispersion of these NPs. In bimetallic catalyst system, Ag–Au alloy NPs in the range of 2–4 nm in diameter were successfully stablized on the surface of the PZS nanotubes (Fig. 3c). Obviously, these alloys didn't aggregate together when the co-reduction was performed, indicating that the PZS nanotubes with abundant hydroxyl groups are a good support for the bimetallic NPs via offering numerous coordinated sites for metal ions. A representative HRTEM image of Ag–Au alloy NPs shows the (111) lattice fringe distance of 0.240 nm (Fig. 3f), which is between the (111) lattice spacing of face-centered cubic (fcc) Ag (0.238 nm, Fig. 3d) and fcc Au (0.241 nm, Fig. 3e) NPs.
 | ||
| Fig. 3 TEM images and size histogram of PZS@Ag NPs composites (a and d), PZS@Au NPs composites (b and e) and PZS@Ag–Au NPs composites (c and f). | ||
PXRD was applied to evaluate the crystalline property of the nanocrystals within the Ag–Au@PZS nanotubes. As displayed in Fig. 4, the pristine PZS nanotubes pattern only contains one broad diffraction band at 2θ value of about 16.8°, ascribed to the amorphous PZS matrix. As for the PZS@Ag NPs composites, the PXRD pattern showed four peaks at 38.2°, 44.3°, 64.5°, and 77.6°, which were indicative of characteristic diffractions due to the face-centered cubic structure of the Ag crystals (JCPDF no. 4-0783). The diffraction pattern of the asprepared PZS@Ag NPs composite are almost the same as that of PZS@Au NPs composite because silver has the similar crystal structure (fcc) and lattice constant as gold (0.409 nm versus 0.408 nm) (JCPDS 4-0783 and 4-0784).42 A further examination of the PZS@Ag–Au NPs composite showed that there was no obvious variation in peak positions, with the exception that sharp peaks at 27.6° and 31.8° were also detected using XRD; they corresponded to the reflections of AgCl precipitation, which formed in the preparation process of bimetallic catalyst.43 It was acceptable because the Ag and Au nanocrystals showed little difference in the PXRD pattern. Therefore, further instruments such as UV-vis spectrometry are needed to examine whether those Au–Ag bimetallic nanoparticles form a homogeneous alloy structure or not.
 | ||
| Fig. 4 PXRD patterns of PZS nanotubes, PZS@Ag NPs composites, PZS@Au NPs composites and PZS@Ag–Au NPs composites. | ||
The UV-vis spectra of PZS@Ag NPs, PZS@Au NPs, PZS@Ag–Au NPs composites are shown in Fig. 5. The absorption bands for the pure PZS@Ag NPs composite and pure PZS@Au NPs composite are at 408 nm and 526 nm, which is consistent with the reported spectra. For Ag–Au bimetallic catalyst, there is only one plasom absorption peak in-between the absorption bands of pure Ag and pure Au in UV-vis spectrum.44,45 This observation indicated that the nanoparticles were an alloy structure with a homogeneous composition, rather than a physical mixture of monometallic Au and Ag nanoparticles. Moreover, when the added amount of HAuCl4 solution (20 mM) was increased from 0.025 mL to 0.2 mL, the broad plasmon resonance peak could be red-shifted gradually. The change in the optical absorption spectra reflected the evolution of the composition and structure of the bimetallic nanocrystals supported on the PZS nanotubes. The broadening of the absorption band of PZS@Ag–Au NPs is because their plasmon resonance combines the energies of both the silver and the gold.46
 | ||
| Fig. 5 The corresponding UV-vis absorption spectra of PZS@Ag NPs composites (i), PZS@Au NPs composites (ii), and PZS@Ag–Au NPs composites synthesized by adding HAuCl4 solution (20 mM) of 0.025 mL (iii), 0.5 mL (iv), 1.0 mL (v), 1.5 mL (vi), and 2.0 mL (vii). | ||
Interesting optical properties are further indicated by the diverse colors of the bimetallic catalyst suspensions (Fig. 6). The Au@PZS nanotubes suspensions are little pink. The colors of the bimetallic composites' suspensions are not merely a mix of the colors of the monometallic ones. The initial bright yellow of PZS@Ag NPs composite turns into a range of colors of PZS@Ag–Au NPs suspensions starting from yellow-green and turning ever more to dusty purple as the more Au precursors are added (see (iii)–(vii) in Fig. 6). The modulation should be attributed to the bimetallic composition and structure.47 It is possible to tune the color over a wide wavelength range by controlling the Ag/Au ratio.
 | ||
| Fig. 6 Images showing the suspensions containing different composite particles. | ||
X-ray photoelectron spectroscopy (XPS) analysis is an effective method to investigate the elemental composition of materials and determine the interaction at the interface. Fig. 7 exhibits the XPS spectra of PZS@Ag NPs composite, PZS@Au NPs composite and PZS@Ag–Au NPs composite. It can be seen from Fig. 7a that Ag, Au nanoparticles do exist in the as-prepared bimetallic catalyst, which is well agreed with the results from the above analysis. The binding energy (BE) of Ag 3d5/2 in the PZS@Ag NPs composite was 368.2 eV, indicating that Ag is in the metallic state (368.0 eV). The binding energy (BE) of Au 4f7/2 for monometallic catalyst PZS@Au NPs composite is 84.3 eV, and the binding energy of Au 4f7/2 in bulk metallic gold is 84.0 eV. The 0.3 eV shift in Au 4f7/2 BE between monometallic catalyst PZS@Au NPs composite and bulk metallic gold may be due to the interaction between the support and Au nanoparticles.48,49 Moreover, for the bimetallic catalysts, Ag 3d5/2 BE and Au 4f7/2 BE have a clear negative shift compared with the monometallic PZS@Ag NPs composite and PZS@Au NPs composite, indicating the presence of a synergistic effect between Ag and Au.
 | ||
| Fig. 7 XPS spectra of the PZS@Ag–Au NPs composites: (a) survey spectrum, (b) Ag 3d spectrum, (c) Au 4f spectrum. | ||
The reduction of 4-nitrophenol (4-NP) by NaBH4 was chosen as a model reaction to characterize the catalytic performance of PZS@Ag–Au NPs composite. The reaction process was monitored by UV-vis spectrometry, as illustrated in Fig. 8. The original 4-NP solution was light yellow in color and showed typical absorption at 319 nm. Upon the addition of NaBH4 into 4-NP solution, the colour quickly changed to yellow green, and the absorption maximum shifted to 400 nm (Fig. 8a) due to the formation of 4-nitrophenolate ions.50 Nevertheless, the absorbance of this peak remained almost unchanged with time in the absence of a catalyst (Fig. 8b).51 With the addition of the 0.05 mg PZS@Au–Ag NPs (entry 6 in Table 1) composite into the reaction mixture, the absorption peak at 400 nm dramatically decreased in intensity, and the yellow-green color faded with the simultaneous gradual development of the new absorption peak at 298 nm, assigned to 4-AP, indicating that the reaction took place in the presence of bimetallic catalyst (Fig. 8c). The catalytic reaction can be estimated to end with the complete disappearance of the UV-vis absorption at 400 nm. Furthermore, we studied the catalytic ability of monometallic PZS@Ag NPs and PZS@Au NPs. All those experiments were carried out under the same conditions and the amount of each catalyst was controlled at 0.05 mg. From the time-dependent UV-vis absorption spectra (Fig. 8d and e), the time for PZS@Ag NPs (50 min) to complete the reaction is similar to that for PZS@Au NPs (55 min), but it is much longer than that for PZS@Ag–Au NPs composite (8 min). This indicated that the catalytic activity of bimetallic PZS@Ag–Au NPs composite is highly enhanced compared to the monometallic composites. Moreover, we studied the catalytic ability of the mixture of PZS@Ag and PZS@Au NPs (1:1 by mass) under the same conditions. As shown in Fig. 8f, the time for the mixture to complete the reaction (45 min) is obviously longer than that for PZS@Ag–Au NPs composite, demonstrating the synergetic effects of Ag and Au in the 4-NP reduction. In addition, the UV-vis spectra showed an isosbestic point (318 nm), suggesting that the catalytic reduction of 4-NP gave only 4-AP, without any other byproduct.52
 | ||
| Fig. 8 UV-vis spectra of 4-NP before and after addition of NaBH4 solution (a), 4-NP with NaBH4 without addition of any catalysts (b), 4-NP with NaBH4 in the presence of PZS@Ag–Au NPs composites as catalysts (c), 4-NP with NaBH4 in the presence of PZS@Ag NPs composites as catalysts (d), 4-NP with NaBH4 in the presence of PZS@Au NPs composites as catalysts (e), the mixture of PZS@Au NPs and PZS@Ag NPs (f). Conditions: 4-NP (1.0 × 10−4 mol L−1, 1.0 mL); NaBH4 (7.5 × 10−3 mol L−1, 1.0 mL); catalyst (0.05 mg). | ||
| Sample | HAuCl4 (mL) | AgNO3 (mL) | Ag (wt%) | Au (wt%) | Au/Ag | k (s−1) | k (s−1 g−1) | |
|---|---|---|---|---|---|---|---|---|
| Weight | Molar | |||||||
| 1 | 0.0 | 0.2 | 1.42 | — | — | — | 0.83 × 10−3 | 16.6 |
| 2 | 0.025 | 0.2 | 1.275 | 0.21 | 0.306 | 0.164 | 1.70 × 10−3 | 34 |
| 3 | 0.05 | 0.2 | 0.912 | 0.35 | 0.701 | 0.384 | 2.47 × 10−3 | 49.4 |
| 4 | 0.1 | 0.2 | 0.703 | 0.57 | 1.48 | 0.81 | 2.92 × 10−3 | 58.4 |
| 5 | 0.15 | 0.2 | 0.43 | 0.62 | 2.63 | 1.44 | 4.41 × 10−3 | 88.2 |
| 6 | 0.2 | 0.2 | 0.35 | 0.74 | 3.91 | 2.14 | 4.61 × 10−3 | 92.2 |
| 7 | 0.2 | 0.0 | — | 1.03 | — | — | 0.378 × 10−3 | 7.56 |
Taking into account the much higher concentration of BH4− than 4-NP, the pseudo-first-order kinetics with respect to 4-NP can be used to evaluate the rate of the catalytic activity.50 The rate constant (k) can be calculated from the rate equation ln(Ct/C0) = −kt, where t is the reaction time, C0 is the initial concentration of 4-NP, and Ct is the concentration of 4-NP at the time t. To optimize the catalytic property of PZS@Ag–Au NPs composite and investigate the synergetic effects of Ag and Au in the 4-NP reduction, we synthesized a series of bimetallic catalyst with different ratios of Au/Ag to compare the variation in their k values. As expected, linear relationships of ln(At/A0) versus reaction time were obtained for all samples. Table 1 presents the variable k values, where the nature of the bimetallic nanocrystals in the composites was tuned by precisely controlling the Au/Ag molar ratios. The percentages of Au and Ag within the PZS@Ag–Au NPs composites were assessed by AAS. PZS@Ag NPs composite and PZS@Au NPs composite were also tested as a control. It is clear that all the PZS@Ag–Au NPs bimetallic catalyst have a higher catalytic activity in comparison with the monometallic catalyst, revealing the synergistic effect of Ag and Au species. Moreover, with increases in the Au content, k accordingly increased, reaching values as high as 4.61 × 10−3 s−1. We also compiled the changing tendency of the k values in Fig. 9, with the aim of determining whether any correlation existed between the catalytic activity and the Au to Ag ratio. When the molar ratio of Au to Ag was varied from 0 to 2.14, the k values first increased drastically and then eventually approached an asymptotic limit. It is most likely that the catalytic activity of the Ag–Au nanocrystals was enhanced with increased Au concentrations in the bimetallic nanocrystals.53 The turnover frequency (TOF), which is defined as the number of molecules formed per active site per second, was calculated to be about 678.9 h−1 for the catalytic reduction of 4-NP by using PZS@Ag–Au NPs composites (entry 6 in Table 1) as a catalyst, which is superior to those noble metal-based catalysts reported in the literature (Table 2). From the above analysis, it can be seen that the PZS@Ag–Au NPs bimetallic catalyst showed the superior catalytic activity, due largely to the nature of bimetallic nanocrystals supported on the PZS nanotubes. This extraordinary performance can be explained by the following two sides. First, structure effects, composite effects, and size effects resulted from the intimate interactions between the two metals plays a key role in determining bimetallic nanomaterials' catalytic performance.54,55 Second, a synergistic electronic effect was existed in the bimetallic nanoparticles: electrons could transfer from Ag to Au, leading to an increase in the electron density on the surface of the bimetallic PZS@Ag–Au nanoparticles, which could improve the catalytic activity.56,57
 | ||
| Fig. 9 Relationship between the catalytic activity parameter (k) and Au/Ag molar ratios when the noble metal nanocrystals supported on PZS nanotubes were transferred from Ag nanocrystals to Au–Ag bimetallic nanocrystals. | ||
To investigate the reusability, the PZS@Ag–Au NPs composites with the highest catalytic activity (entry 6 in Table 1) were used in the reduction reaction of 4-NP, over six cycles. From Fig. 10, it was found that the conversion of the 4-nitrophenol could still reach 87.9% after six cycles, indicating that the obtained PZS@Ag–Au NPs composites exhibited a good reusability. We are aware of the slight decrease in conversion, which should be due to the loss of the nanocatalysts after each cycles and the adsorption of the 4-aminophenol on the surface of noble metal nanoparticles.58
 | ||
| Fig. 10 Reusability of the PZS@Ag–Au NPs composites (entry 6 in Table 1) as catalysts for the reduction of 4-NP by NaBH4. | ||
Conclusions
In summary, we have demonstrated a very simple and rapid method to efficiently fabricate PZS@Ag–Au NPs composites. The presence of heteroatoms and a large number of hydroxyl groups in PZS nanotubes could enhance surface wettability and facilitate the obtainment of ultra-fine bimetallic NPs with good dispersion and small particle size (about 2.6 nm). By tuning the added amounts of the Ag and Au precursor, a series of PZS@Ag–Au NPs composites with different Au-to-Ag molar ratios were synthesized, among which bimetallic catalysts exhibited extraordinary catalytic activity compared to its monometallic counterparts. We also observed the catalytic capability is remarkably enhanced when the Au contents is increased; the activity parameter showed values as high as 92.2 s−1 g−1, which suggests a significant dependence of the synergistic effect on the ratios of the components of the bimetallic catalysts. More importantly, the catalytic capability has retained ∼86% of the initial activity after reuse for 6 cycles. We believe that such bimetallic composites could have great potential applications in the future. It is also expected that this facile and effective approach could be extended to fabricate many kinds of other high performance bimetallic NPs on appropriate supporting materials, which generate new features and applications.Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 51003098, 21101141, 51473149), the Foundation of State Key Laboratory of Chemical Engineering (No. SKL-ChE-13A04), the National Science Foundation for Post-doctoral Scientists of China (No. 2014M550385), the Foundation of Henan Educational Committee for Key Program of Science and Technology (No. 14A430026), the Foundation of Zhengzhou General Science and Technology Project (No. 141PPTGG385), and the financial support from the Program for New Century Excellent Talents in Universities (NCET).References
- M. H. Huang, S. Mao, H. Feick, H. Q. Yan, Y. Y. Wu, H. Kind, E. Weber, R. Russo and P. D. Yang, Science, 2001, 292, 1897–1899 CrossRef CAS PubMed.
- B. T. Holland, C. F. Blanford and A. Stein, Science, 1998, 281, 538–540 CrossRef CAS PubMed.
- J. Z. Zhang, A. M. Schwartzberg, T. Norman, C. D. Grant, J. Liu, F. Bridges and T. van Buuren, Nano Lett., 2005, 5, 809–810 CrossRef CAS PubMed.
- P. Veerakumar, R. Madhu, S. M. Chen, V. Veeramani, C. T. Hung, P. H. Tang, C. B. Wang and S. B. Liu, J. Mater. Chem. A., 2014, 2, 16015–16022 CAS.
- M. Nasrollahzadeh, S. M. Sajadi, A. R. Vartooni and M. Bagherzadeh, J. Colloid Interface Sci., 2015, 448, 106–113 CrossRef CAS PubMed.
- S. C. Tang, S. Vongehr and X. K. Meng, J. Mater. Chem., 2012, 20, 5436–5445 RSC.
- Z. C. Xu, Y. L. Hou and S. H. Sun, J. Am. Chem. Soc., 2007, 129, 8698–8699 CrossRef CAS PubMed.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- Y. G. Sun and Y. N. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS PubMed.
- J. Zeng, Q. Zhang, J. Y. Chen and Y. Xia, Nano Lett., 2010, 10, 30–35 CrossRef CAS PubMed.
- S. C. Warren, F. J. Disalvo and U. Wiesner, Nat. Mater., 2007, 6, 156–161 CrossRef CAS PubMed.
- U. Banin, Nat. Mater., 2007, 6, 625–626 CrossRef CAS PubMed.
- Q. Zhang, X. Guo, Z. X. Liang, J. H. Zeng, J. Yang and S. J. Liao, Nano. Res., 2013, 6, 571–580 CrossRef CAS.
- S. Biswas, C. O. Regan, M. A. Morris and J. D. Holmes, Small, 2015, 11, 103–111 CrossRef CAS PubMed.
- D. S. Wang and Y. D. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
- Y. H. Dai, Y. Wang, B. Liu and Y. H. Yang, Small, 2015, 11, 268–289 CrossRef CAS PubMed.
- M. X. Li, J. T. Hu, Z. X. Chen and H. B. Lu, RSC Adv., 2014, 4, 41152–41158 RSC.
- Q. S. Liu, Z. Yan, N. L. Henderson, J. C. Bauer, D. W. Goodman, J. D. Batteas and R. E. Schaak, J. Am. Chem. Soc., 2009, 131, 5720–5721 CrossRef CAS PubMed.
- J. W. Zhang, C. P. Hou, H. Huang, L. Zhang, Z. Y. Jiang, G. X. Chen, Y. Y. Jia, Q. Kuang, Z. X. Xie and L. S. Zheng, Small, 2013, 9, 538–544 CrossRef CAS PubMed.
- D. S. Wang and Y. D. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
- S. Y. Song, R. L. Liu, Y. Zhang, J. Feng, D. P. Liu, Y. Xing, F. Y. Zhao and H. J. Zhang, Chem.–Eur. J., 2010, 16, 6251–6256 CrossRef CAS PubMed.
- X. K. Wang, Q. G. Liu, Z. H. Xiao, X. Chen, C. Shi, S. Y. Tao, Y. Q. Huang and C. H. Liang, RSC Adv., 2014, 4, 48254–48259 RSC.
- S. C. Tang, S. Vongehr and X. K. Meng, J. Phys. Chem. C, 2010, 114, 977–982 CAS.
- S. Sahoo, S. Husale, S. Karna, S. K. Nayak and P. M. Ajayan, J. Am. Chem. Soc., 2011, 133, 4005–4009 CrossRef CAS PubMed.
- G. Y. Yang, S. Shao, Y. H. Ke, C. L. Liu, H. F. Ren and W. S. Dong, RSC Adv., 2015, 5, 37112–37118 RSC.
- Y. Chi, J. C. Tu, M. G. Wang, X. T. Li and Z. K. Zhao, J. Colloid Interface Sci., 2014, 423, 54–59 CrossRef CAS PubMed.
- K. Kondo, T. Kida, Y. Ogawa, Y. Arikawa and M. Akashi, J. Am. Chem. Soc., 2010, 132, 8236–8237 CrossRef CAS PubMed.
- Z. C. Wang, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 15954–15955 CrossRef CAS PubMed.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1444 CrossRef CAS PubMed.
- M. Steinhart, J. H. Wendorff, A. Greiner, R. B. Wehrspohn, K. Nielsch, J. Schilling, J. Choi and U. Gosele, Science, 2002, 296, 1997–1999 CrossRef CAS PubMed.
- S. Ko and J. Jang, Angew. Chem., Int. Ed., 2006, 45, 7564–7567 CrossRef CAS PubMed.
- B. Ding, J. Gong, J. Kim and S. Shiratori, Nanotechnology, 2005, 16, 785–790 CrossRef CAS.
- R. K. Zheng, Y. Yang, Y. Wang, J. Wang, H. Chan, C. L. Choy, C. G. Jin and X. G. Li, Chem. Commun., 2005, 1447–1449 RSC.
- H. R. Allcock, M. S. Connolly, J. T. Sisko and S. Al-Shali, Macromolecules, 1988, 21, 323–334 CrossRef CAS.
- M. Gleria, A. Bolognesi, W. Porzio, M. Catellani, S. Destri and G. Audisio, Macromolecules, 1987, 20, 469–473 CrossRef CAS.
- H. R. Allcock, C. J. Nelson and W. D. Coggio, Organometallics, 1991, 10, 3819–3825 CrossRef CAS.
- H. Bai, C. Li and G. Q. Shi, Adv. Mater., 2011, 23, 1089–1115 CrossRef CAS PubMed.
- G. Goncalves, P. A. A. P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Gracio, Chem. Mater., 2009, 21, 4796–4802 CrossRef CAS.
- L. Zhu, Y. Y. Xu, W. Z. Yuan, J. Y. Xi, X. B. Huang, X. Z. Tang and S. X. Zheng, Adv. Mater., 2006, 18, 2997–3000 CrossRef CAS.
- X. Z. Wang, J. W. Fu, M. H. Wang, Y. J. Wang, Z. M. Chen, J. N. Zhang, J. F. Chen and Q. Xu, J. Mater. Sci., 2014, 49, 5056–5065 CrossRef CAS.
- M. H. Wang, J. W. Fu, D. D. Huang, C. Zhang and Q. Xu, Nanoscale, 2013, 5, 7913–7919 RSC.
- Y. Wang, J. M. Zheng, K. N. Fan and W. L. Dai, Green Chem., 2011, 13, 1644 RSC.
- R. Adhikari, G. Gyawali, T. Sekino and S. W. Lee, J. Solid State Chem., 2013, 197, 560–565 CrossRef CAS.
- M. P. Mallin and C. J. Murphy, Nano Lett., 2002, 2, 1235–1237 CrossRef CAS.
- T. Wu, L. Zhang, J. P. Gao, Y. Liu, C. J. Gao and J. Yan, J. Mater. Chem. A, 2013, 1, 7384–7390 CAS.
- T. Y. Olson, A. M. Schwartzberg, C. A. Orme, C. E. Talley, B. O'Connell and J. Z. Zhang, J. Phys. Chem. C, 2008, 112, 6319–6329 CAS.
- A. M. Schwartzberg, T. Y. Olson, C. E. Talley and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 19935–19944 CrossRef CAS PubMed.
- P. Zhang, C. L. Shao, Z. Y. Zhang, M. Y. Zhang, J. B. Mu, Z. C. Guo, Y. Y. Sun and Y. C. Liu, Nanoscale, 2011, 3, 17746–17753 Search PubMed.
- J. Li, C. Y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426–8430 RSC.
- S. Jana, S. K. Ghosh, S. Nath, S. Pande, S. Praharaj, S. Panigrahi, S. Basu, T. Endo and T. Pal, Appl. Catal., A, 2006, 313, 41–48 CrossRef CAS.
- J. M. Song, S. S. Zhang and S. H. Yu, Small, 2014, 10, 717–724 CrossRef CAS PubMed.
- Y. H. Deng, Y. Cai, Z. K. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Y. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS PubMed.
- Q. An, M. Yu, Y. T. Zhang, W. F. Ma, J. Guo and C. C. Wang, J. Phys. Chem. C, 2012, 116, 22432–22440 CAS.
- Y. H. Feng, W. P. Xue, H. B. Yin, M. J. Meng, A. L. Wang and S. X. Liu, RSC Adv., 2015, 5, 106918–106929 RSC.
- X. H. Peng, Q. M. Pan and G. L. Rempel, Chem. Soc. Rev., 2008, 37, 1619–1628 RSC.
- X. M. Chen, Z. X. Cai, X. Chen and M. Oyama, J. Mater. Chem. A, 2014, 2, 5668–5674 CAS.
- B. H. Xia, F. He and L. D. Li, Langmuir, 2013, 29, 4901–4907 CrossRef CAS PubMed.
- P. Wang, L. Han, C. Z. Zhu, Y. M. Zhai and S. J. Dong, Nano. Res., 2011, 4, 1153–1162 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |