
A nanocomposite based on multi-walled carbon nanotubes grafted by molecularly imprinted poly(methacrylic acid–hemin) as a peroxidase-like catalyst for biomimetic sensing of acetaminophen
Ederson dos Santos Morettia,
Juliana de Fátima Giarolab,
Michele Kucekia,
Maiyara Carolyne Pretea,
Arnaldo César Pereirab and
César Ricardo Teixeira Tarley*ac
aUniversidade Estadual de Londrina (UEL), Departamento de Química, Centro de Ciências Exatas, Rodovia Celso Garcia Cid, PR 445, km 380, Londrina, PR 86050-482, Brazil. E-mail: tarley@uel.br; Fax: +55 43 3371 4286; Tel: +55 43 3371 4366
bDepartamento de Ciências Naturais, Universidade Federal de São João del Rei (UFSJ), Campus Dom Bosco, Praça Dom Helvécio 74, Fábricas, 36301-160, São João del Rei, MG, Brazil
cInstituto Nacional de Ciência e Tecnologia (INCT) de Bioanalítica, Universidade Estadual de Campinas (UNICAMP), Instituto de Química, Departamento de Química Analítica, Cidade Universitária Zeferino Vaz, s/n, Campinas, SP 13083-970, Brazil
First published on 11th March 2016
Abstract
In the present study the synthesis of a nanocomposite based on multi-walled carbon nanotubes grafted by poly(methacrylic acid–hemin) is described. The presence of functional groups in the nanocomposite was evaluated by FT-IR, while morphological, textural and thermal stability data were evaluated by means of SEM, TEM, nitrogen adsorption–desorption assays and TGA, respectively. The material was evaluated as a biomimetic and catalytic sensor toward the electrochemical determination of acetaminophen (AC) by depositing it onto a glassy carbon electrode surface. Acetaminophen has been widely used as an analgesic and antipyretics drug and for patients with a high sensitivity to aspirin; therefore the development of analytical methods for AC determination in pharmaceutical formulations is of paramount importance for quality control. The biomimetic electrochemical sensor operation is similar to that of the biosensor based on horseradish peroxidase, where the hemin in the presence of H2O2 catalyses the oxidation of acetaminophen into N-acetyl-p-benzoquinoneimine, which in turn is reduced back to AC on the electrode at a potential of −0.27 V. The variables that exert influence on the performance of the biomimetic response, including H2O2 concentration (300 μmol L−1), pH (8.0) and concentration (0.1 mol L−1) and type of buffer solution (Trizma) were investigated. Under optimized conditions the electrochemical determination of AC using multi-walled carbon nanotubes grafted by poly(methacrylic acid–hemin) immobilized on the surface of the glassy carbon electrode was carried out by square wave voltammetry (SWV), showing a limit of detection of 1.1 μmol L−1. The selectivity of material, ascribed to imprinted effect, was assured by comparison to a non-imprinted material (NIP). The proposed method was applied to AC determination in a pharmaceutical formulation, whose obtained values were very similar to those declared and statistically equal to the HPLC method, thus illustrating the feasibility of method for analysis of real samples.
1. Introduction
The reliable determination of drugs in bulk and pharmaceutical formulations is of paramount importance for quality control to prevent overdose while optimizing the individual dosage regimen.1,2 N-Acetyl-p-aminophenol (paracetamol or acetaminophen, AC) belongs to analgesic and antipyretics drugs and has been used extensively in the world to moderate pain and can be an alternative drug for patients with high sensitivity to aspirin. However, it has been reported that acute overdoses of AC may cause renal insufficiency due to accumulation of toxic metabolites resulting in death of patient.3,4 Although many analytical techniques are available for the determination of AC in pharmaceutical formulation, including titrimetry,5 high performance liquid chromatography,6 and UV-vis spectrophotometry,7 extensive efforts have been made for the development of electroanalytical methods, owing to their high simplicity, low-cost, sensibility and rapid analysis.8 In general, when bare carbon electrodes, such as glassy carbon or graphite paste electrodes are used, the electrooxidation of AC takes place in high overpotential, which in turn decreases the selectivity of electrochemical measurements. In addition, these electrodes can suffer from fouling by oxidation and interfering products formed and usually require periodic mechanical polishing or activation processes, which decreases the sample throughput of analytical procedure. These drawbacks have been successfully overcome by preparation of biosensors using horseradish peroxidase as catalyst element immobilized on nanocomposite carbon nanotubes/conductor polymer,9 entrapped in graphite paste10,11 or immobilized on core–shell ZrO@Fe3O4 nanoparticles on chitosan hybrid film electrodeposited on the surface of an Au electrode.1 In spite of biosensors offer intrinsic selectivity toward the substrate, their successful use depends upon the properties of the native enzyme, and after repeated measurements their catalytic activity is decreased.12 In this sense, molecularly imprinted polymers (MIP) and biocatalyst (non-enzymatic system) based on synthetic metallic complex have proven to be interesting biomimetic materials with substantial stability over biosensors, but with similar selectivity.13,14 Molecularly imprinted polymers are synthesized in the presence of a molecule template, which establishes interactions with the functional monomer. After polymerization, the template is removed from the polymeric matrix, thereby resulting in the formation of analyte-selective binding sites.15The usefulness of MIP as platform in electrochemical sensing depends on its adequate integration on the surface of transducer electrode and some strategies have been reported for this task. Yola et al. developed a new voltammetric sensor based on Fe@Au nanoparticles involved in 2-aminoethanethiol functionalized multi wall carbon nanotubes for the determination of cefexima in human plasma.16 The combined use of nanoparticles, nanocarbonaceous material and molecularly imprinted polymer has been also reported. Yola et al. described the electrochemical polymerization of phenol as monomer on substrate of silver nanoparticles (AgNPs) involved in a polyoxometalate (H3PW12O40, POM) functionalized reduced graphene oxide (rGO) deposited on the surface of glassy carbon electrode. The sensor was applied to ochratoxin A determination in wine and grape juice.17 A similar study was developed using platinum nanoparticles (PtNPs) and the sensor was applied to citrinin (CIT) determination in rye samples.18 Electropolymerization of MIP on the surface of glassy carbon electrode is another strategy for the preparation of based-MIP electrochemical sensor as reported by Gupta et al.19 In this study, a molecularly imprinted polypyrrole (PPy) was fabricated for the determination of tobramycin (TOB) in egg and milk. Molecularly imprinted technology has been very effective to generate sensitive quartz crystal microbalance (QCM). For instance, in the study reported by Gupta et al.20 the development of sensor is based on modification of gold surface of QCM chip through self-assembly monolayer formation with allyl mercaptane, with further polymerization of poly(2-hydroxyethylmethacrylate–methacryloylamidoaspartic acid) [p(HEMA–MAAsp)] film in the presence of template kaempferol (KAE). The feasibility of sensor was evaluated by analysis of orange and apple juices for the determination of KAE. In this direction, this strategy has been successfully employed for the determination of tobramycin (TOB) in pharmaceuticals and foods20 and lovastatin (LOV) in red yeast rice.21 The strategy of template polymerization on the gold surface modified with ally mercaptane is also very useful for preparation of surface plasmon resonance (SPR) sensor.22,23 As observed, some strategies for the preparation of based-MIP sensor have been reported, but a brief survey of literature reveals that few studies dedicated to the use of MIP as a electrochemical sensing device for AC determination have been noticed and one of the most common strategy involves the electrochemical synthesis of conductive MIP onto bare electrode (glassy carbon electrode and graphite electrode).24,25 For instance, Teng et al. developed a layer of conductive film of molecularly imprinted poly(p-aminobenzene sulfonic acid) (pABSA) on the surface of glassy carbon electrode for the determination of AC.26 In a similar study, Luo et al. designed the synthesis of a molecularly imprinted conductive polyaniline (PANI) nanoparticles using polymeric micelle as nanoreactors electrochemically deposited onto glassy carbon electrode for the determination of AC by differential pulse voltammetry.27 Although the developed conductive MIP presents high selectivity toward the AC owing to the imprinting effect created during polymer synthesis; on the other hand, the electrooxidation peak of AC still took place in high overpotential. Thus, the incorporation of a catalyst element in the synthesis of MIP, such as metalloporphyrins as both co-monomer and catalytic center is an interesting way to improve the performance of MIP for electrochemical purposes, and thus, the MIP acts as an artificial enzyme.28,29 Furthermore, the use of conductive MIP is limited due to the low commercial availability of conductive monomers. Therefore, the synthesis of non-conductive catalytic MIP grafted on the surface of carbon nanotubes (CNT) by covalent linkages seems to be a very promising approach for obtaining MIP as an enzyme-mimicking catalyst for the electrochemical determination of AC and other phenolic compounds. The use of CNT in the nanocomposite may provide high electrical conductivity to the material, ensure higher dispersibility of polymer and the nanoscale of MIP grafted on the surface of CNT improves the mass transfer of analyte towards the selective binding sites, unlike the MIP synthesized by bulk method.30,31
According to aforementioned, the aim of the current study is to synthesize a catalytic nanocomposite and evaluate its use as a new electrochemical sensing platform for AC, combining the intrinsic properties of CNT, the merits of molecular imprinting and the catalytic properties of Fe(III)protoporphyrin(IX) (hemin). The synthesized nanocomposite was based on multi-walled carbon nanotubes grafted by poly(methacrylic acid–hemin) and used as an enzyme-mimicking catalyst. The material was characterized by FT-IR, TGA, SEM and TEM and the electrode performance for real analysis was checked from the electrochemical analysis of pharmaceutical formulations.
2. Experimental
2.1. Apparatus
Voltammetric measurements were performed at room temperature, with a potentiostat/galvanostat Autolab® PGSTAT-12 (Eco Chemie BV, Utrecht, The Netherlands). An electrochemical cell containing a glassy carbon electrode (3 mm) modified with nanocomposite as the working electrode, an Ag/AgCl electrode as the reference and a platinum wire as the auxiliary electrode were used. The voltammetric measurements were carried out in the absence of oxygen, by bubbling N2 gas. In order to evaluate the morphology of material, a microscope JEOL® JSM-6360 LV equipped with dispersive energy microscopy was used. Before analysis, the material was coated with a thin layer of gold, using a Bal-Tec MED 020 equipment, in order to minimize charging under the incident electron beam. The FT-IR spectra were recorded in the 4000–400 cm−1 region with a spectrophotometer model 8300 Shimazdu (Tokyo, Japan), using the KBr pellet conventional method. Thermal stability of materials was evaluated from thermogravimetric analysis (TGA) using a TGA 4000 Perkin Elmer (Waltham, USA) thermobalance. For analysis, ∼10 mg of sample was heated from 30 to 900 °C at a rate of 10 °C min−1, under nitrogen flow of 20 mL min−1. The transmission electron microscopy images were taken using a microscope JEOL® JEM-1400 with an accelerating voltage of 120 kV. The samples were previously dispersed in ethanol by sonication for 20 min and then the suspension was dripped on copper grids and dried under vacuum. The pH measurements were performed using pH meter Metrohm® 827 (Herisau, Switzerland). Chromatography determination was performed on a Shimadzu liquid chromatograph equipped with a LC-20AT gradient pump, a UV-VIS diode array detector at 225.0 nm and an injector fitted with a 20 μL loop. Acetaminophen separation was carried out on a Shimadzu CLC-ODS (M) column (column size: 250 mm × 4.6 mm i.d., particle size: 5 μm) at 25 °C operating at a flow rate of 1.0 mL min−1 using a methanol–phosphate buffer 0.1 mol L−1 (pH = 6.2) as mobile phase (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, v/v).2
:50, v/v).2
2.2. Reagents
Acetaminophen (AC, Sigma-Aldrich, >98%), hydroquinone (Sigma-Aldrich, ≥99%), catechol (Sigma-Aldrich, ≥98%), L-DOPA (Sigma-Aldrich, >98%), ascorbic acid (Sigma-Aldrich, 99%), uric acid (Sigma-Aldrich, >98%), methacrylic acid (MAA, Sigma-Aldrich, 99.5%), trimethylolpropane trimethacrylate (TRIM, Sigma-Aldrich, 90%), 2,2′-azoisobutyronitrile (AIBN, Sigma-Aldrich, 98%), vinyltrimethoxysilane (VTMS, Sigma-Aldrich, ≥97.5%), Fe(III)protoporphyrin(IX) (Sigma-Aldrich, ≥90%), toluene (HPLC grade ≥ 99.9%), dimethyformamide (DMF, Sigma-Aldrich, ≥99.8%), hydroquinone (HQ, Sigma-Aldrich, ≥99%), Nafion® (Sigma-Aldrich, 5%), sodium acetate (Sigma-Aldrich, ≥99%), HEPES (Sigma-Aldrich, ≥99.5%), sodium phosphate (Sigma-Aldrich, 96%), Trizma (Sigma-Aldrich, ≥98.5%), methanol (Sigma-Aldrich, 99.5%), acetic acid (Sigma-Aldrich, 99.5%), H2SO4 (Sigma-Aldrich, 95–95%), HNO3 (Sigma-Aldrich, >90%), H2O2 (Sigma-Aldrich, 29–32%) and MWCNTs, supplied by CNTs Co. Ltd. Yeonsu-Gu, Incheon, Korea 93% 10–40 nm diameter and length of 5–20 μm. All the solutions were prepared in deionized water from a water purification system Milli-Q® (Bedford, USA).2.3. Synthesis of catalytic nanocomposite
Firstly, MWCNT was oxidized according to literature data.32 300 mg of MWCNT were mixed with 40.0 mL of a HNO3/H2SO4 (3:1, v/v) mixture and refluxed at 65 °C for 2 h. Afterwards, the material was successively washed with deionized water (pH ≈ 7.0) to remove acid excess, dried at 60 °C and stored at room temperature. After oxidation process, 150 mg of oxidized MWCNT were dispersed in 30.0 mL of toluene in a three-neck round-bottom flask and kept in an ultrasonic bath (40 kHz) for 20 min. In the next step, 3.6 g of vinyltrimetoxisilane previously dissolved in 20.0 mL of toluene containing 1% (m/m) of hydroquinone was slowly added to the mixture under stirring. One of the three necks was sealed, the central neck was coupled to a reflux condenser and the other neck was purged with nitrogen (N2) for 10 min. Afterwards, the neck was sealed and the mixture was heated at 100 °C in an oil bath for 6 h. The obtained functionalized MWCNT was washed with ethanol and dried at 50 °C for 6 h. For the synthesis of catalytic nanocomposite, 40.0 mg of functionalized MWCNT were dispersed in 20.0 mL of toluene in a flat-bottomed flask and kept in an ultrasonic bath (40 kHz) for 20 min. Then, 0.5 g of methacrylic acid (5.8 mmol) as functional monomer, 0.45 g of trimethylolpropane trimethacrylate (1.32 mmol) as cross-linking reagent, 60 mg of Fe(III)protoporphyrin(IX) (0.097 mmol), as prosthetic group of peroxidase and 45.35 mg of AC as template molecule were added to the flask. The mixture was manually stirred, followed by addition of 80.0 mg of 2,2′-azoisobutyronitrile (0.48 mmol). Nitrogen gas was purged for 5 min and then the flask was sealed. The mixture was incubated for 24 h at 60 °C under stirring. The obtained catalytic nanocomposite was then washed with methanol/acetic acid (4:1, v/v) for the removal of acetaminophen, dried at 60 °C for 12 h and stored in amber flask until use. The obtained material was named as MIP. The blank polymer, i.e., the non imprinted polymer (NIP) was synthesized in similar way excepted by the addition of AC. The schematic representation of synthesis of nanocomposite based on multi-walled carbon nanotubes grafted by poly(methacrylic acid–hemin) is depicted in Fig. 1.
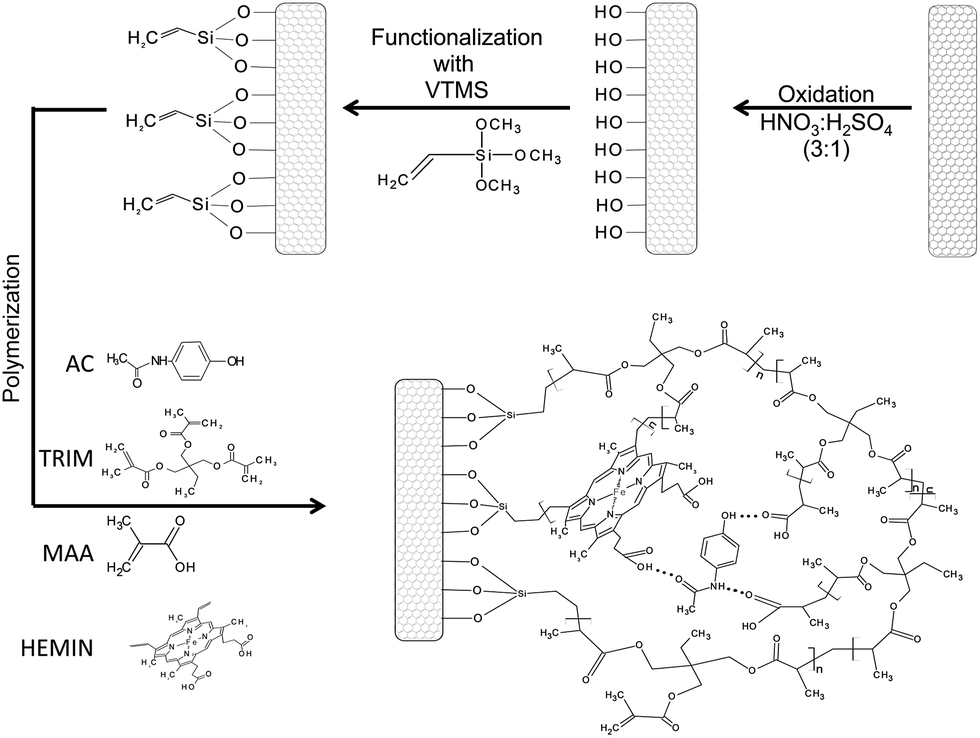 | ||
| Fig. 1 Schematic representation of synthesis of nanocomposite based on multi-walled carbon nanotubes grafted by poly(methacrylic acid–hemin). | ||
2.4. Electrochemical procedure
The nanocomposite was cast on the surface of a glassy carbon electrode by pipetting 10.0 μL of sonicated suspension of 10.0 mg mL−1 nanocomposite dispersed in DMF followed by addition of 10.0 μL of 5% (m/v) Nafion® and kept under heating at 45 °C in an oven to volatilize the solvent. The measurements of square wave voltammetry (SWV) were carried out in a conventional electrochemical cell with 15.0 mL capacity containing the modified glassy carbon electrode, Ag/AgCl (KCl, 3.0 mol L−1) reference electrode and the platinum auxiliary electrode containing 0.1 mol L−1 Trizma buffer at pH 8.0 and using frequency of 10 Hz with pulse amplitude of 20 mV.2.5. Sample preparation
Five tablets with known declared content (750 mg per tablet) were ground to a fine powder and thoroughly mixed. Then, a known mass was weighed for the sample and diluted with 5.0 mL of water and sonicated during 10 min.33 The same procedure was adopted to another sample containing 500 mg per tablet. Afterwards, the solution was transferred to a volumetric flask whose volume was made up to 100.0 mL and a volume of 20 μL of solution was transferred into the electrochemical cell containing 0.1 mol L−1 Trizma buffer at pH 8.0 and the volume was made up to 10.0 mL.3. Results and discussion
3.1. Characterization of MWCNT and nanocomposite
The FT-IR spectra of materials are depicted in Fig. 2. For the pristine MWCNT, oxidized MWCNT and MWCNT functionalized with vinyltrimethoxysilane (Fig. 2a), the intense absorption band observed at 3450 cm−1 can be attributed to the ν(O–H) stretching vibrations on the MWCNT surface and adsorbed water that forms hydrogen bonds.32,34 A carbonyl band at 1630 cm−1 was noticed35 and at 1380 cm−1 the low intense band is attributed to δ(O–H) related to in-plane vibration of carboxylic acid.36 The stretching of C–O from primary alcohol was observed at 1090 cm−1, while, the low intensity signals at 770 and 660 are ascribed to the Si–O stretching vibrations, which confirms the surface functionalization of MWCNT with vinyltrimethoxysilane.37 After polymerization with MIP and NIP, the spectra profiles were somewhat different (Fig. 2b). The broad signal in the region of 2990–2950 cm−1 is ascribed to symmetric and asymmetric CH2 stretching vibrations from the backbone chain of polymer. The occurrence of a well defined band at 1726 cm−1 was observed, which can be assigned to stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from MAA or hemin. In this case, the shoulder at 1632 cm−1 can be attributed to CC stretching from vinyl groups or overlap by OH deformation (water).38 The signals at 1260 and 1160 cm−1 can be attributed to C–N from porphyrinic backbone of hemin or stretching of C–O from ester group and carboxylic acid.39 The signals at 1470 and 1390 cm−1 can be ascribed, respectively, to C–H from methylene and methyl groups.38 The similarity of MIP and NIP spectra also confirms the complete removal of AC from polymeric matrix of MIP using methanol/acetic acid (4:1, v/v) solution.
O from MAA or hemin. In this case, the shoulder at 1632 cm−1 can be attributed to CC stretching from vinyl groups or overlap by OH deformation (water).38 The signals at 1260 and 1160 cm−1 can be attributed to C–N from porphyrinic backbone of hemin or stretching of C–O from ester group and carboxylic acid.39 The signals at 1470 and 1390 cm−1 can be ascribed, respectively, to C–H from methylene and methyl groups.38 The similarity of MIP and NIP spectra also confirms the complete removal of AC from polymeric matrix of MIP using methanol/acetic acid (4:1, v/v) solution.
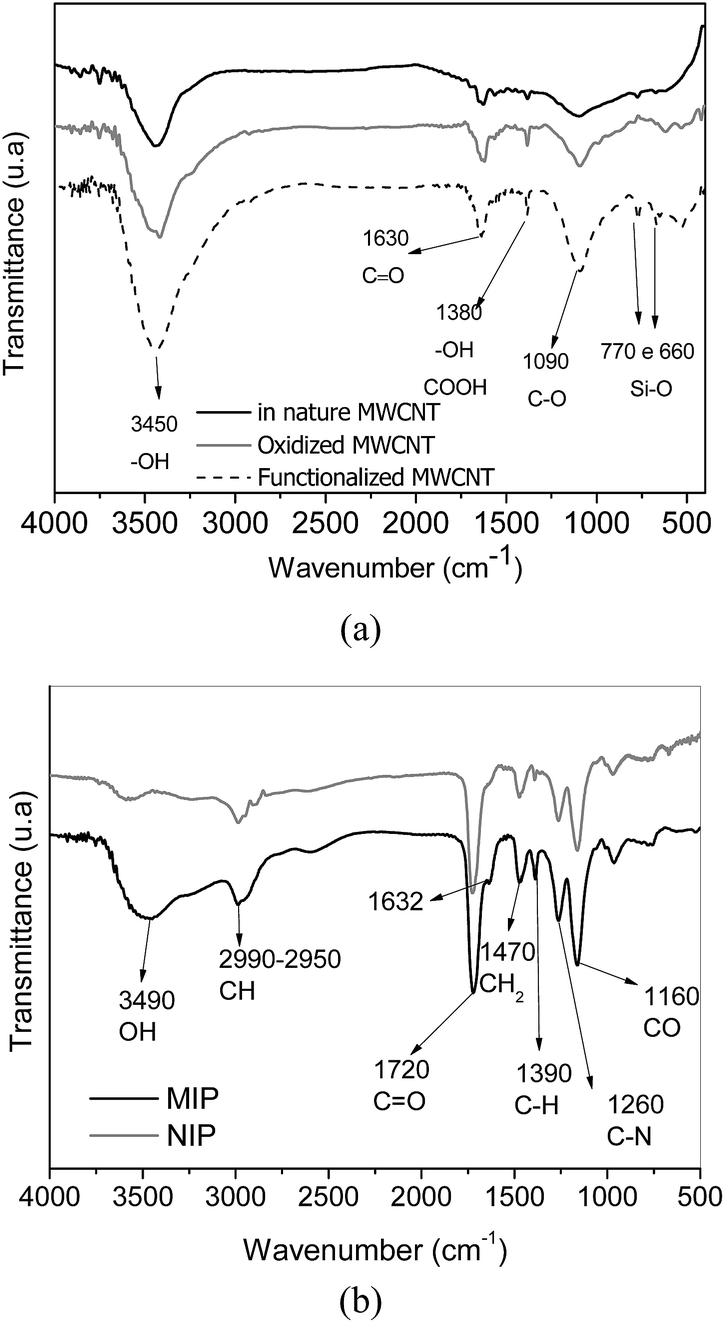 | ||
| Fig. 2 FT-IR spectra for the materials. (a) MWCNT, oxidized MWCNT and functionalized MWCNT, (b) MIP and NIP. | ||
Fig. 3a and b shows the thermogravimetric curves of pristine MWCNT, oxidized MWCNT, MWCNT functionalized with vinyltrimethoxysilane, MIP and NIP. As can be observed, the thermal profile of the MWCNTs are very similar to each other, with weight loss of 17% at 900 °C, which demonstrates that polymerization reaction of vinyltrimethoxysilane resulting in inorganic matrix was not observed. On the other hand, differences in thermal profiles between MIP and NIP were evident regarding the MWCNTs. The first weight loss of 9% occurs in the range of 30–90 °C, which is ascribed to the volatilization of adsorbed water. Weight loss of 25% for MIP and NIP occurs, respectively, at 390 °C and 350 °C, thus indicating that the former one is thermally more stable than NIP. Upon temperature range of 300–470 °C the decomposition of segments of the polymeric chain of methacrylic acid and cross-linking reagent trimethylolpropane trimethacrylate takes place.38 Taking into account the temperature range varying from 200 to 470 °C for MIP and NIP, it can be observed a weight loss of 78%, i.e., much higher than that achieved for MWCNT. Such difference in the weight loss indicates the amount of polymer grafted onto MWCNT surface of the total weight of nanocomposite.
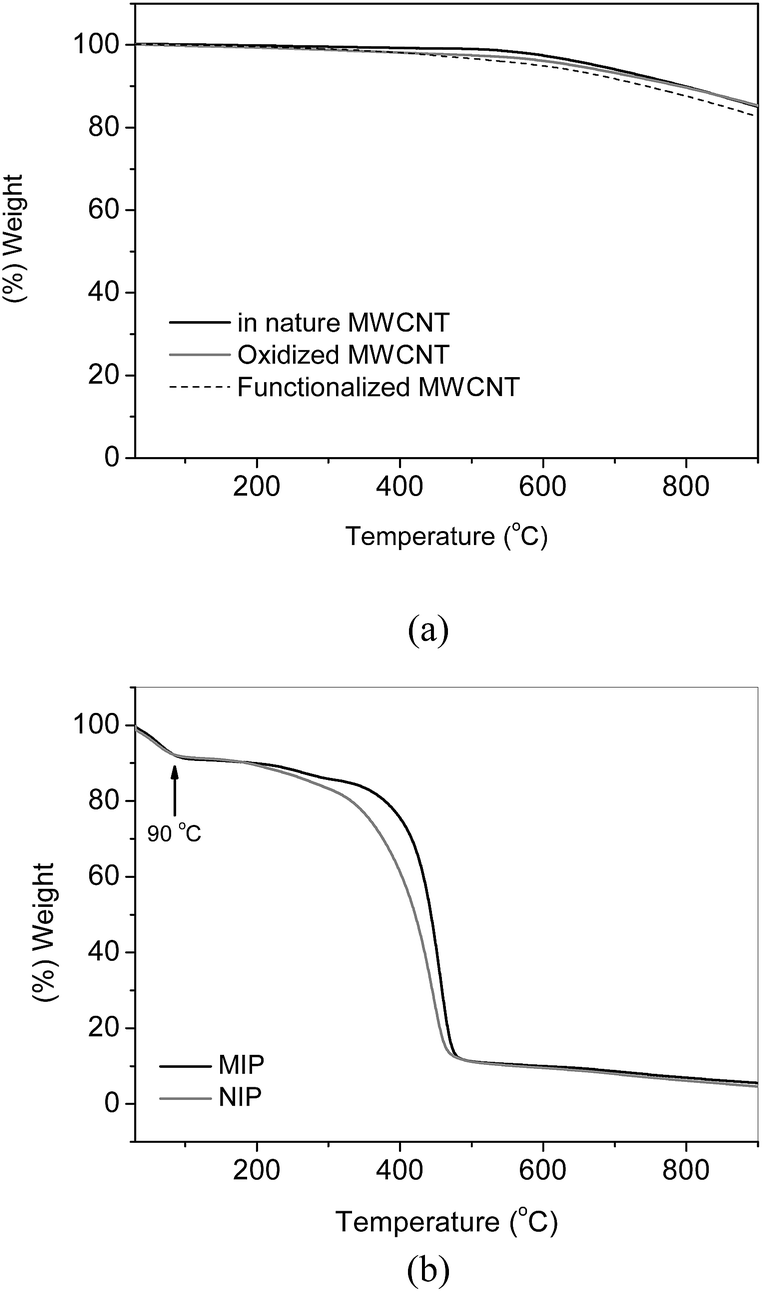 | ||
| Fig. 3 TG curves for the materials. (a) MWCNT, oxidized MWCNT and functionalized MWCNT, (b) MIP and NIP. | ||
The SEM images of pristine MWCNT, oxidized MWCNT, functionalized MWCNT, MIP and NIP are depicted in Fig. 3. The morphological characteristics of MWCNT are observed in Fig. 3a, with entangled filamentous of carbon nanotubes. The oxidation of MWCNT, as well as the silanization reaction with vinyltrimetoxisilane, promote intense aggregation of the carbon nanotubes filaments (Fig. 4b and c). After grafting process of polymers on the surface of MWCNT, it is possible to clearly observe an increase in the diameter of MWCNT, thus indicating that the MWCNTs are densely covered by polymers (Fig. 4d and e). From TEM images (Fig. 5) this finding is distinctly observed, where the polymeric matrix is homogeneously distributed over the surface of MWCNT in nanoscale (10–50 nm).
 | ||
| Fig. 4 SEM images for the materials. (a) MWCNT, (b) oxidized MWCNT, (c) functionalized MWCNT, (d) MIP and (e) NIP. Magnification 30000×. | ||
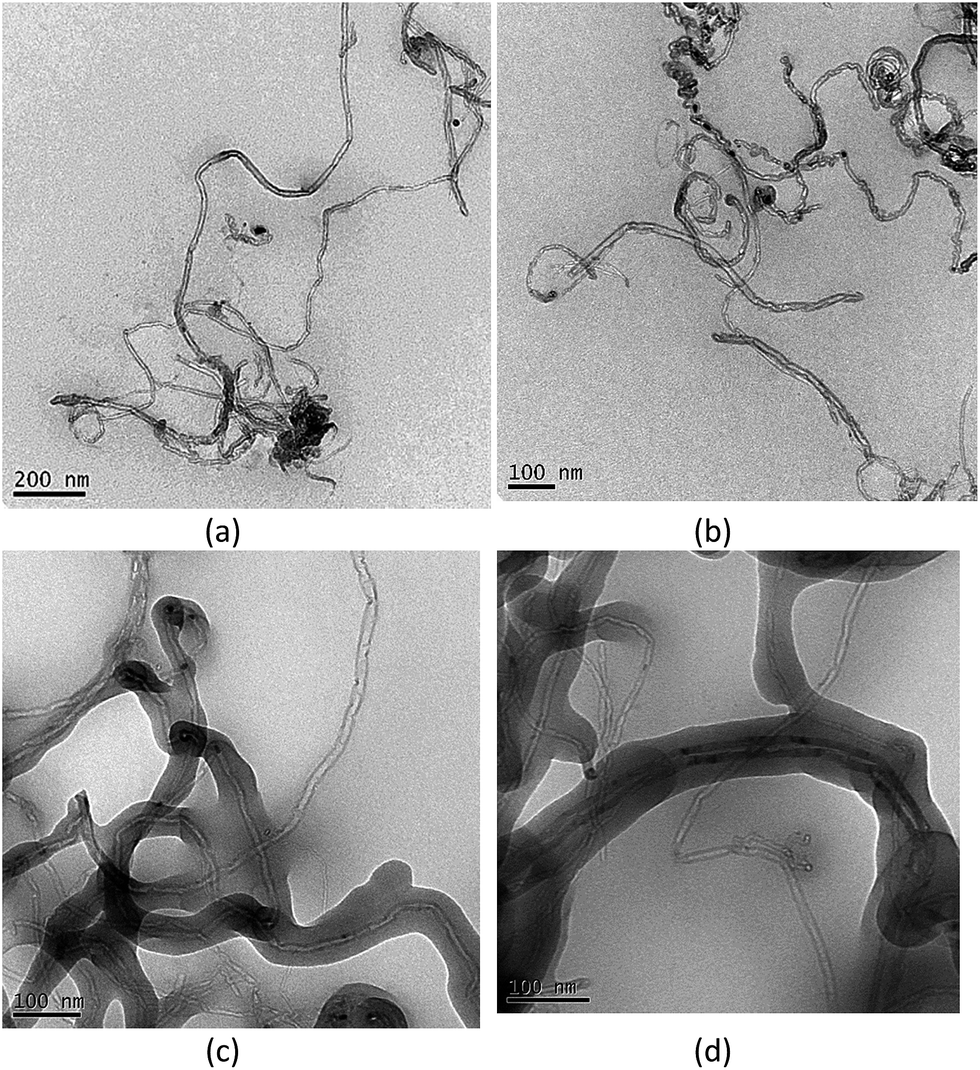 | ||
| Fig. 5 TEM images for the materials. (a and b) Functionalized MWCNT, (c and d) MIP. | ||
The textural data, including surface area, pores volume and pores diameter of nanocomposite, play an important role in the electroanalytical performance toward the AC determination. Table 1 shows the differences in textural data between MIP and NIP, where the porosity of NIP was higher than that achieved for MIP. This result can be attributed to the greater solubility of complex template (AC)-functional monomers (MAA–hemin) in porogenic solvent during the synthesis of MIP, which reduced the solvent removal rate from polymeric matrix and, as a consequence, decreased its porosity.40 The lower porosity of MIP compared to NIP indicates in advance that differences in the electrochemical behavior of modified sensor will be attributed to the imprinted cavities created in the MIP, and not to the textural data dependence.
| Nanocomposites | Surface area (m2 g−1) | Pores volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| MIP | 13.84 | 0.037 | 3.4 |
| NIP | 22.37 | 0.083 | 15.9 |
3.2. Electrochemical behavior of MIP and NIP nanocomposites toward the AC determination
Fig. 6a presents the cyclic voltammetric curves obtained from MIP nanocomposite electrode in 0.1 mol L−1 phosphate buffer solution at pH 7.0. The assays were carried out to evaluate the properties of MIP as a peroxidase-like catalyst for biomimetic sensing of AC using cathodic scan from 0.4 to −0.5 V. In the absence of H2O2 and AC, it was observed a reduction peak at −0.2 V corresponding to the Fe(III)/Fe(II) redox couple of iron present in the hemin, in similar way to the redox couple of the heme protein of horseradish peroxidase.41 After adding H2O2, the cathodic peak current was increased, thus showing that hemin exhibits, as expected, catalytic properties toward the H2O2 reduction in the same way as peroxidase-based biosensors. In the presence of H2O2, the addition of AC promotes a significant increase in the peak current, which demonstrates that the hemin in the presence of H2O2 catalyses the chemical oxidation of acetaminophen and the reaction product, i.e., N-acetyl-p-benzoquinoneimine, may in turn be electrochemically reduced on the surface of electrode at lower potential (−0.27 V). | ||
| Fig. 6 Cyclic voltammograms of the MIP nanocomposite electrode (a), bare glassy carbon electrode (b) and (c) NIP nanocomposite electrode in 0.1 mol L−1 phosphate buffer solution, pH 7.0 at 30 mV s−1 in the presence of H2O2 and H2O2 with AC. | ||
In order to verify the influence of MIP nanocomposite toward the AC determination, cyclic voltamograms were recorded using a glassy carbon electrode without any modification. Fig. 6b shows the absence of cathodic peak current in the presence of H2O2 and AC, revealing a better response for the MIP nanocomposite and its catalytic properties.
The better ability of MIP regarding NIP to recognize the AC can be observed by comparing the cyclic voltammograms recorded in Fig. 6a and c. As observed in Fig. 6c, the presence of hemin in the polymeric matrix of NIP makes possible to catalyze the reduction of H2O2 as previously verified for MIP (Fig. 6a). However, only a slight increase of cathodic peak current was observed after adding AC, which clearly proves that the stronger affinity of MIP nanocomposite toward the AC is attributed to the specific binding sites.
3.3. Effect of number of layers and Nafion® concentration on the sensor preparation
A set of assays were carried out in order to define the best conditions for the sensor preparation. Firstly, the number of layers (1 up to 3 layers) of MIP nanocomposite suspension at 10 mg L−1 was evaluated by pipetting 10.0 μL of the suspension, followed by addition of 10.0 μL of 5% (m/v) Nafion®. Cyclic voltammograms for the concentration of 50 μmol L−1 AC in a 0.1 mol L−1 phosphate buffer solution, pH 7.0 and at 30 mV s−1 were recorded. The results showed that only one layer was sufficient to efficiently modify the surface of glassy carbon electrode and provide catalytic properties to the sensor. On the other hand, a larger number of layers decreases the peak current of AC most likely due to blocking of electron flow. Therefore, the casting of glassy carbon electrode was carried out with one layer. It has been reported that Nafion® film has an important role in fixing the nanocomposite layer on the electrode surface. Hence, it was necessary to evaluate the influence of Nafion® concentration in the sensor preparation. Concentrations of 0.5, 1.0, 1.6, 3.0 and 5.0% (m/v) were assessed. It was found that peak current for AC was very similar for all concentrations; however, the highest stability of MIP nanocomposite was achieved by using 5% (m/v) Nafion®, therefore this concentration was selected for further experiments.3.4. Effect of scan rate on the AC electrooxidation
In order to assess the nature of electron transfer process of AC at the MIP nanocomposite, cyclic voltammograms for concentration of 70 μmol L−1 AC in 0.1 mol L−1 phosphate buffer solution, pH 7.0 at different scan rate (20–100 mV s−1) were recorded (Fig. 7).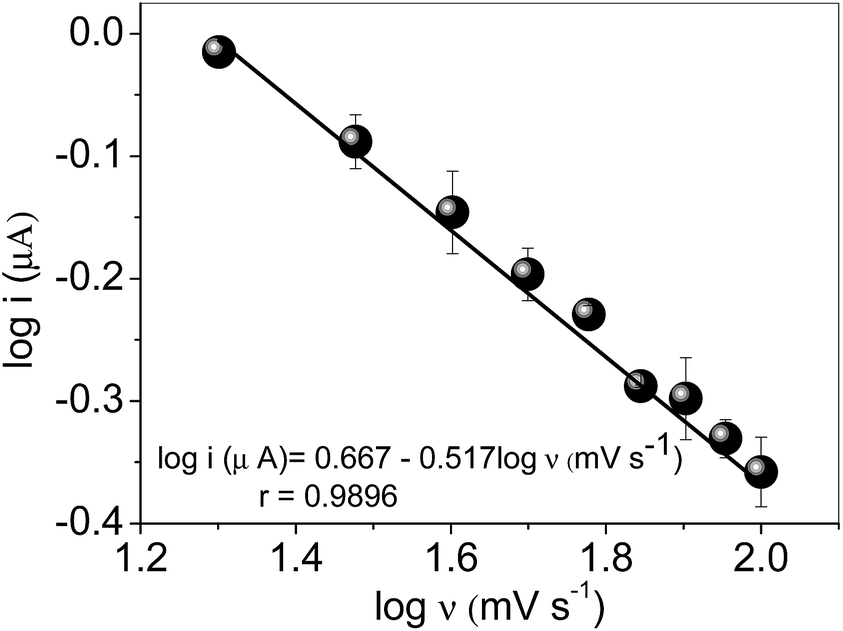 | ||
| Fig. 7 Plot logν versus logi. Cyclic voltammograms recorded in 0.1 mol L−1 phosphate buffer solution, pH 7.0 in the presence of 70 μmol L−1 AC. | ||
As verified in the Fig. 7, the slope (0.517) is very close to the theoretical expected value of 0.5, which confirms that electrochemical reaction of AC takes place through diffusion-controlled process.2
3.5. Effect of pH and H2O2 concentration
The pH of samples has an indicative role for MIP recognition containing hemin as catalytic center in a similar way to heme protein of horseradish peroxidase. Therefore, as shown in Fig. 8, the pH dependence was investigated in the range of 6.0–8.0, to obtain the best condition for the horseradish peroxidase.28 In the 6.0–7.5 pH range, there is no significant difference in the peak current. However, a higher signal was noticed at pH 8.0, so this value was chosen for subsequent work. This finding can be rationalized bearing in mind that the pKa value of AC is 9.5, which in turn makes easier its interaction with specific binding sites.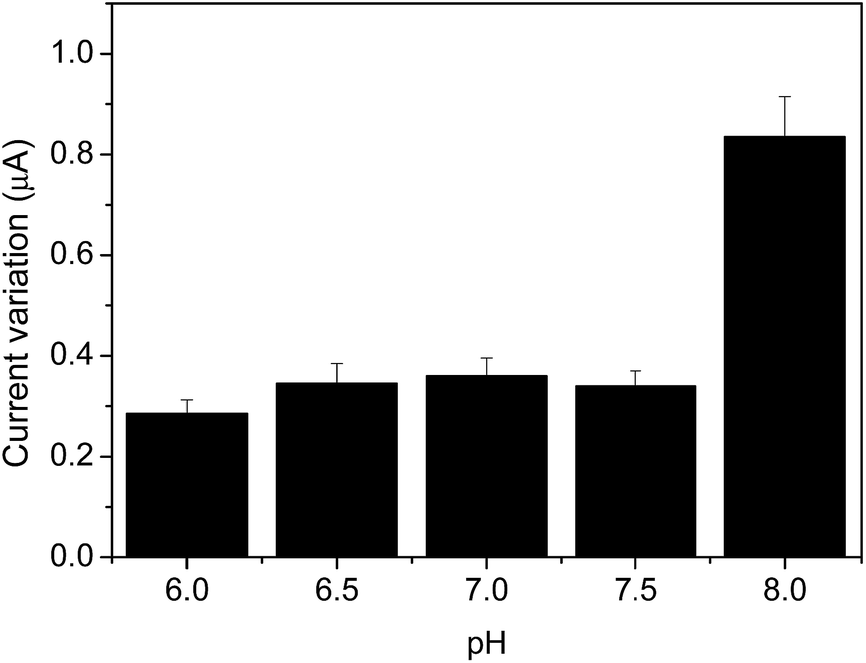 | ||
| Fig. 8 Effect of pH on sensor performance. Conditions: cyclic voltammograms obtained from the MIP nanocomposite electrode in 0.1 mol L−1 phosphate buffer solution, at 30 mV s−1 in the presence of 10.0 μmol L−1 AC. | ||
The H2O2 concentration ranging from 25.0–500 μmol L−1 was also investigated (Fig. 9).
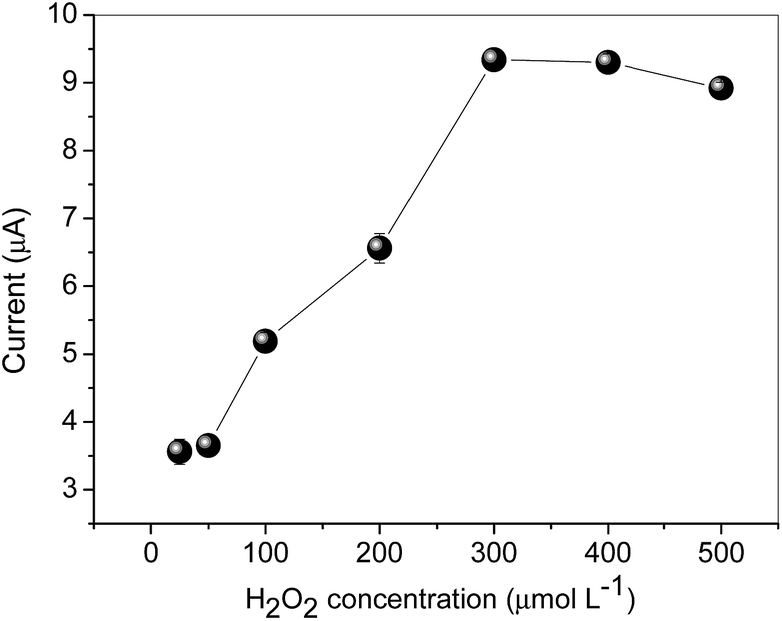 | ||
| Fig. 9 Effect of H2O2 concentration on sensor performance. Conditions: cyclic voltammograms obtained from the MIP nanocomposite electrode in 0.1 mol L−1 phosphate buffer solution, at pH 8.0 and 30.0 mV s−1 in the presence of 10.0 μmol L−1 AC. | ||
As shown in Fig. 9, the maximum peak current was obtained using 300.0 μmol L−1 H2O2, and after this value, a slight decrease in current was observed, which is attributed to the saturation of hemin. Thus, 300.0 μmol L−1 H2O2 solution was selected for further studies.
After obtaining the best conditions for pH and H2O2 concentration, the effect of type of supporting electrolyte was evaluated. Acetate, Hepes, phosphate and Trizma buffer solutions at 0.1 mol L−1 concentrations were evaluated as supporting electrolytes. The assays were carried out by cyclic voltammograms obtained from the MIP nanocomposite electrode at pH 8.0, 30.0 mV s−1, in the presence of 300.0 μmol L−1 H2O2 and increasing concentration of AC (10.0, 30.0, 50.0 and 70.0 μmol L−1). The best result in terms of sensibility (A L μmol−1) was found using Trizma; therefore, the subsequent experiments were carried out by fixing this buffer solution as supporting electrolyte.
3.6. Response of MIP and NIP nanocomposite sensors to other compounds
The selectivity of MIP and NIP nanocomposite sensors was evaluated from the analysis of their response toward other compounds. Some of the compounds are structurally similar to AC and all of them present electrochemical activity. The study was carried out by cyclic voltammetry under pH 7.0 at 30.0 mV s−1 in the presence of 50.0 μmol L−1 of H2O2 and 10.0 μmol L−1 of each compound. As can be seen in Table 2, the developed electrochemical sensor shows excellent response toward the AC when compared to other electroactive molecules. Moreover, higher currents for catechol and hydroquinone were achieved for the NIP nanocomposite in detriment of MIP nanocomposite. These results confirm once again the imprinting effect created during the polymer synthesis.| Compound | Molecular structure | Current cathodic (μA) | Response percentage (%) | ||
|---|---|---|---|---|---|
| MIP | NIP | MIP | NIP | ||
| Acetaminophen |  |
1.05 | 0.07 | 100 | 100 |
| Hydroquinone |  |
0.14 | 0.14 | 13.8 | 200 |
| Catechol |  |
0.07 | 0.14 | 7 | 200 |
| L-DOPA |  |
0 | 0 | 0 | 0 |
| Ascorbic acid |  |
0 | 0 | 0 | 0 |
| Uric acid |  |
0 | 0 | 0 | 0 |
3.7. Choice of electroanalytical technique and figures of merit
From previous studies, the best parameters obtained for SWV were 10 Hz, pulse amplitude of 20 mV and potential step of 4 mV, while for DPV (differential pulse voltammetry) the best values were 20 mV for pulse amplitude, 30 mV s−1 for scan rate and pulse time of 170 ms. Under these conditions, the analytical curves, linear range, limits of detection, determined according to IUPAC recommendation42 and sensitivity summarized in Table 3 were obtained. According to achieved results, SWV has shown better figures of merit for AC determination and has been employed for determining AC in pharmaceutical formulations.| Analytical technique | Linear range (μmol−1) | Analytical curve | LD (μmol−1) | S (A L μmol−1) |
|---|---|---|---|---|
| a LD = limit of detection, S = sensitivity. | ||||
| SWV | 10.0–130.0 | I (μA) = 0.70 + 0.010[AC, μmol−1) (r = 0.995) | 2.7 | 0.010 |
| DPV | 10.0–90.0 | I (μA) = 1.8 + 0.028[AC, μmol−1) (r = 0.991) | 1.1 | 0.028 |
For assessing the precision of proposed method, standard solutions of 30.0 and 90.0 μmol−1 AC were subjected to 10 intra-day measurements by SWV under optimized conditions. Relative standard deviation (RSD) values of 7.2 and 2.7% were found for respective concentrations of 30.0 and 90.0 μmol−1 AC, thus demonstrating the precision of method. Regarding the stability of MIP nanocomposite sensor, it was observed that 60 measures performed in two different days may be performed using 90.0 μmol−1 AC without decreases of analytical performance, yielding RSD value of 4.5%.
3.8. Analytical application of proposed method in real sample
The feasibility of method was assessed through analysis of pharmaceutical formulation. As shown in Table 4, good agreement of achieved results with those described by manufacturer was noticed, which indicates the absence of matrices effect. Moreover, the accuracy of proposed method was checked by comparison with HPLC data, used as a reference technique. No statistical difference was observed between proposed method (paired student t test, at the confidence level of 95%, tcalc = 0.97 lower than ttab = 12.70)43 and HPLC method.| Sample | Labeled value (mg per tablet) | Sensora (mg per tablet) | HPLCa | E1 (%) | E2 (%) |
|---|---|---|---|---|---|
| a Results are expressed as mean value ± standard deviation based on three replicates (n = 3). E1 = sensor vs. HPLC; E2 = sensor vs. labeled value. | |||||
| 1 | 500 | 471.5(±0.5) | 472.2(±5.0) | −0.1 | 5.6 |
| 2 | 750 | 771.4(±0.5) | 722.9(±3.5) | 6.3 | 3.6 |
The accuracy of method was also verified by addition and recovery tests. Table 5 shows that values of recovery are close to 100% for the proposed method and HPLC, thus attesting once again that components in the matrix did not affect the performance of the voltammetric sensor and the efficiency of sample treatment.
| Sample | Labeled value (mg per tablet) | Added AC (mg per tablet) | Sensora (mg per tablet) | HPLCa | R1 (%) | R2 (%) |
|---|---|---|---|---|---|---|
| a Results are expressed as mean value ± standard deviation based on three replicates (n = 3). R1 = recovery percentage by sensor; R2 = recovery by HPLC. | ||||||
| 1 | 500 | — | 471.5(±0.5) | 472.2(±5.0) | — | — |
| 200 | 671.5(±0.67) | 657.1(±5.8) | 100 | 92.4 | ||
| 2 | 750 | 771.4(±0.5) | 722.9(±3.5) | — | — | |
| 200 | 975.1(±0.5) | 916.2(±4.3) | 101.8 | 96.6 | ||
4. Conclusions
The present study describes for the first time the development of a new electrochemical sensing platform for AC determination based on nanocomposite multi-walled carbon nanotubes grafted by molecularly imprinted poly(methacrylic acid–hemin) as a peroxidase-like catalyst. From the FT-IR, TG, SEM and TEM data, it was found that the synthesis and the grafting process of catalytic MIP at nanoscale level on the surface of MWCNT was successfully carried out. The use of surface molecular imprinting technique onto MWCNT for the preparation of catalytic nanocomposite MIP provided a better recognition capacity for AC regarding NIP, as well as better selectivity in the presence of other phenolic compounds, thus showing to be a useful platform to obtain high selective electrochemical sensors. Regarding the analytical characteristics of the sensor, it was found similar limits of detection when compared to other reported electrochemical methods based on molecularly imprinted technology25,44,45 and biomimetic sensor.46 Nevertheless, one should note that the great highlight of proposed method relies on the synergic effect of MIP as a recognition element, the catalytic properties of hemin and conductive properties of MWCNT, thus allowing the AC determination at very low potential.Acknowledgements
The authors would like to thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Project No. 481669/2013-2, 305552/2013-9, 472670/2012-3), Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES) (25/2014), Fundação Araucária do Paraná (163/2014), Laboratório de Espectroscopia da Central de Multiusuário da PROPPG, SANEPAR, FAPEMIG and Instituto Nacional de Ciência e Tecnologia de Bioanalítica (INCT) (Project No. 573672/2008-3) for their financial support and fellowships.References
- J. Narang, N. Malhotra, S. Singh, G. Singh and C. S. Pundir, RSC Adv., 2015, 5, 2396 RSC.
- E. H. Duarte, L. T. Kubota and C. R. T. Tarley, Electroanalysis, 2012, 24, 2291 CrossRef CAS.
- P. A. Vaughan, L. D. L. Scott and J. F. McAleer, Anal. Chim. Acta, 1991, 248, 361 CrossRef CAS.
- R. T. Kachoosangi, G. G. Wildgoose and R. G. Compton, Anal. Chim. Acta, 2008, 618, 54 CrossRef CAS PubMed.
- G. Burgot, F. Auffret and J. L. Burgot, Anal. Chim. Acta, 1997, 343, 125 CrossRef CAS.
- E. McEvoy, S. Donegan, J. Power and K. Altria, J. Pharm. Biomed. Anal., 2007, 44, 137 CrossRef CAS PubMed.
- M. L. Ramos, J. F. Tyson and D. J. Curran, Anal. Chim. Acta, 1998, 364, 107 CrossRef CAS.
- N. Havensa, P. Trihna, D. Kimb, M. Lunab, A. K. Wanekayab and A. Mugwerua, Electrochim. Acta, 2010, 55, 2186 CrossRef.
- M. Tertis, A. Florea, R. Sandulescu and C. Cristea, Sensors, 2013, 13, 4841 CrossRef CAS PubMed.
- I. C. Vieira, K. O. Lupetti and O. F. Filho, Quim. Nova, 2003, 26, 39 CrossRef CAS.
- A. Erdem, A. Pabuccuoulu, B. Meruc, K. Kerman and M. Ozsos, Turk. J. Med. Sci., 2000, 30, 349 CAS.
- Y. Hasebe, T. Akiyama, T. Yagisawa and S. Uchiyama, Talanta, 1998, 47, 1139 CrossRef CAS PubMed.
- M. R. S. Ruy, E. C. Figueira and M. D. P. T. Sotomayor, J. Braz. Chem. Soc., 2015, 26, 2069 Search PubMed.
- T. Huynh and W. Kutner, Biosens. Bioelectron., 2015, 74, 856 CrossRef CAS PubMed.
- D. N. Clausen, I. M. R. Pires and C. R. T. Tarley, Mater. Sci. Eng., C, 2014, 44, 99 CrossRef CAS PubMed.
- M. L. Yola, T. Eren and N. Atar, Biosens. Bioelectron., 2014, 60, 277 CrossRef CAS PubMed.
- M. L. Yola, V. K. Gupta and N. Atar, Mater. Sci. Eng., C, 2016, 61, 368 CrossRef CAS PubMed.
- N. Atar, M. L. Yola and T. Eren, Appl. Surf. Sci., 2016, 362, 315 CrossRef CAS.
- V. K. Gupta, M. L. Yola, N. Özaltin, N. Atar, Z. Üstündag and L. Uzun, Electrochim. Acta, 2013, 112, 37 CrossRef CAS.
- V. K. Gupta, M. L. Yola and N. Atar, Sens. Actuators, B, 2014, 194, 79 CrossRef CAS.
- M. L. Yola, L. Uzun, N. Özaltin and A. Denizli, Talanta, 2014, 120, 318 CrossRef CAS PubMed.
- M. L. Yola, N. Atar and T. Eren, Sens. Actuators, 2014, 198, 70 CrossRef CAS.
- N. Atar, T. Eren and M. L. Yola, Food Chem., 2015, 184, 7 CrossRef CAS PubMed.
- J. Luo, C. Fan, X. Wang, R. Liu and X. Liu, Sens. Actuators, B, 2013, 188, 909 CrossRef CAS.
- L. Özcan and Y. Sahin, Sens. Actuators, B, 2007, 127, 362 CrossRef.
- T. Teng, L. Fan, Y. Dai, M. Zhong, X. Lu and X. Kan, Biosens. Bioelectron., 2015, 71, 137 CrossRef PubMed.
- J. Luo, J. Sun, J. Huang and X. Liu, Chem. Eng. J., 2016, 283, 1118 CrossRef CAS.
- L. R. Sartori, W. J. R. Santos, L. T. Kubota, M. G. Segatelli and C. R. T. Tarley, Mater. Sci. Eng., C, 2011, 31, 114 CrossRef CAS.
- R. Santos, P. R. Lima, C. R. T. Tarley, N. F. Hoehr and L. T. Kubota, Anal. Chim. Acta, 2009, 2, 170 CrossRef PubMed.
- H. Ghadimi, R. M. A. Tehrani, A. S. M. Ali, N. Mohamed and S. A. Ghani, Anal. Chim. Acta, 2013, 765, 70 CrossRef CAS PubMed.
- S. Hong, M. Kim, C. K. Hong, D. Jung and S. E. Shim, Synth. Met., 2008, 158, 900 CrossRef CAS.
- M. Z. Corazza, B. F. Somera, M. G. Segatelli and C. R. T. Tarley, J. Hazard. Mater., 2012, 243, 326 CrossRef CAS PubMed.
- Y. Umasankar, B. Unnikrishnan, S. M. Chen and T. W. Ting, Int. J. Electrochem. Sci., 2012, 7, 484–498 CAS.
- N. B. Wutke, K. M. Diniz, M. Z. Corazza, F. M. de Oliveira, E. S. Ribeiro, B. T. da Fonseca, M. G. Segatelli and C. R. T. Tarley, Anal. Lett., 2016, 49, 723 CrossRef CAS.
- N. Chopra, M. Majumder and B. J. Hinds, Adv. Funct. Mater., 2005, 15, 858 CrossRef CAS.
- J. Coates, Interpretation of infrared spectra, a practical approach, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, John Wiley & Sons, Chichester, UK, 2000, pp. 10815–10837 Search PubMed.
- B. F. Somera, M. Z. Corazza, M. J. S. Yabe, M. G. Segatelli, E. Galunin and C. R. T. Tarley, Water, Air, Soil Pollut., 2012, 223, 6069–6081 CrossRef CAS.
- R. J. da Fonseca, M. G. Segatelli, K. B. Borges and C. R. T. Tarley, React. Funct. Polym., 2015, 93, 1–9 CrossRef CAS.
- T. Yeuk-Ki, H. Yee-Man and L. K. Sze-Yin, Talanta, 2012, 89, 162–168 CrossRef PubMed.
- T. Renkecz, G. Mistlberger, M. Pawlak, V. Horváth and E. Bakker, ACS Appl. Mater. Interfaces, 2013, 3, 8537–8545 Search PubMed.
- X. Liu, H. Feng, J. Zhang, R. Zhao, X. Liu and D. K. Y. Wong, Biosens. Bioelectron., 2012, 32, 118–194 CrossRef PubMed.
- G. L. Long and J. D. Winefordner, Anal. Chem., 1983, 55, 712–724 CrossRef.
- B. B. Neto, I. S. Scarminio and R. E. Bruns, Como fazer Experimentos: Aplicações na Ciência e na Indústria, Bookman, São Paulo, 4th edn, 2010 Search PubMed.
- C. Malitesta, I. Losito and P. G. Zambonin, Anal. Chem., 1999, 71, 1366 CrossRef CAS PubMed.
- A. Gómez-Caballero, M. A. Goicolea and R. J. Barrio, Analyst, 2005, 130, 1012 RSC.
- M. C. Q. Oliveira, M. R. V. Lanz, A. A. Tanakac and M. D. P. T. Sotomayor, Anal. Methods, 2010, 2, 507 RSC.
| This journal is © The Royal Society of Chemistry 2016 |