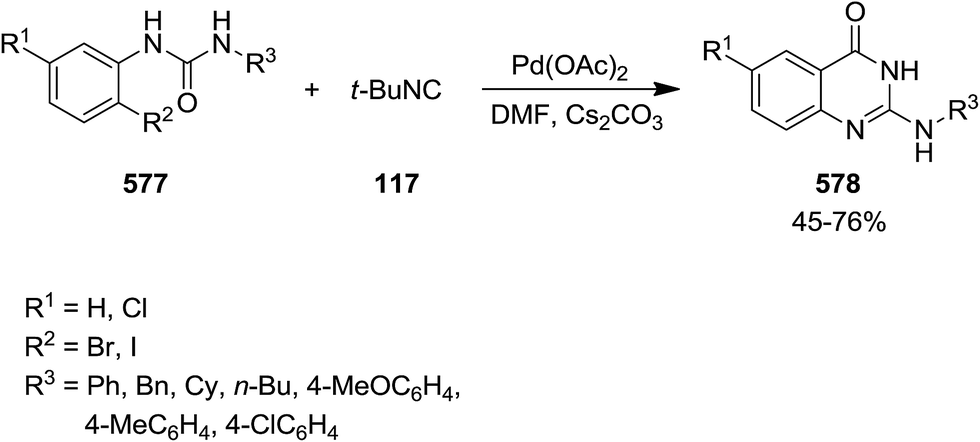
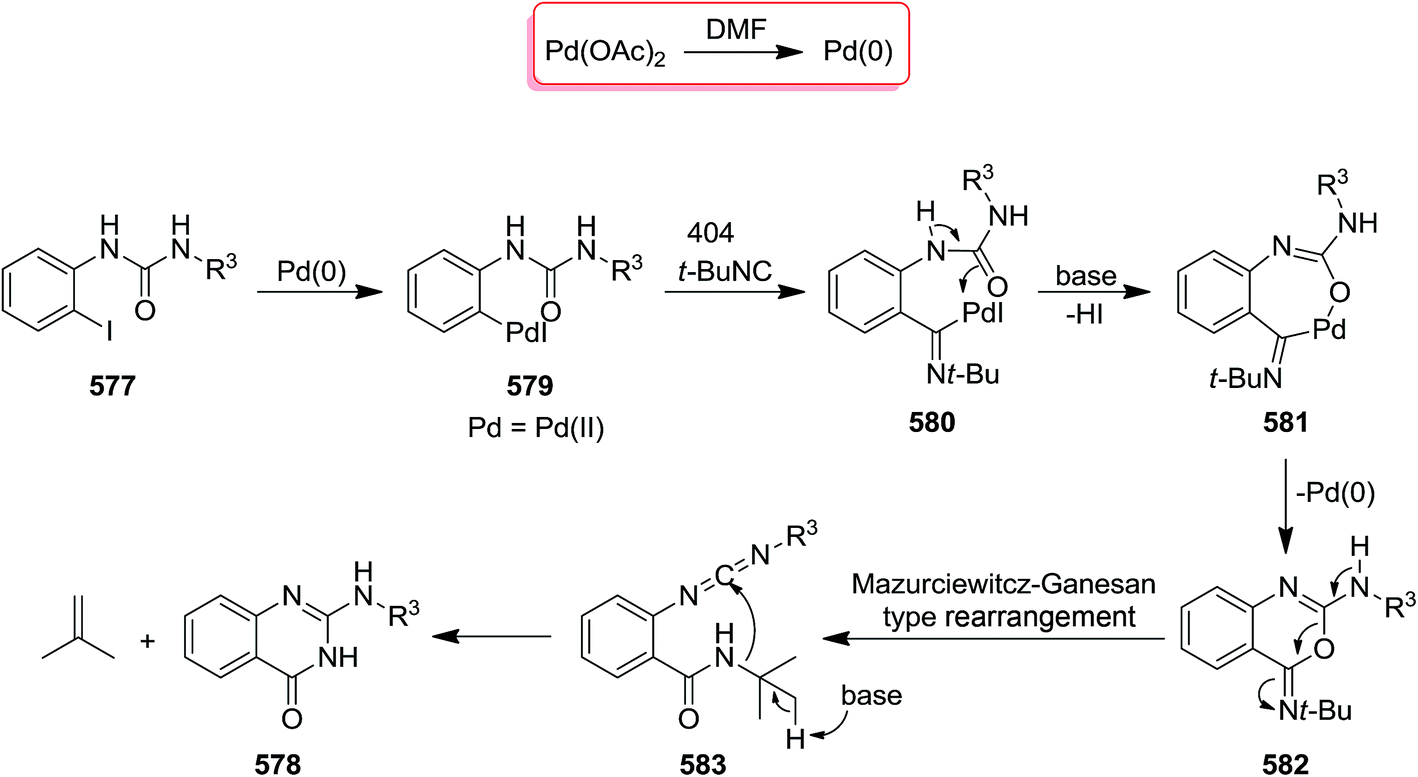
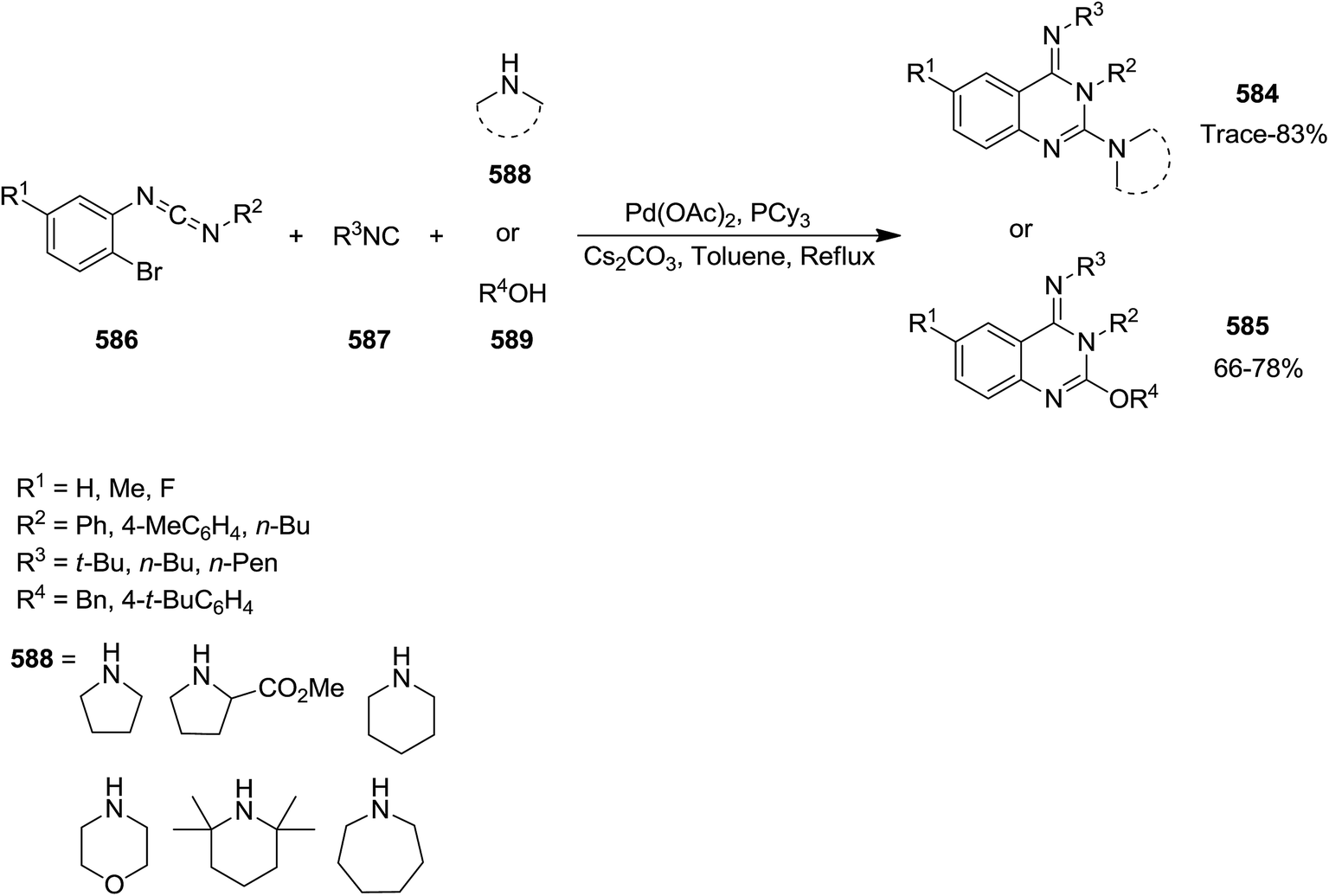
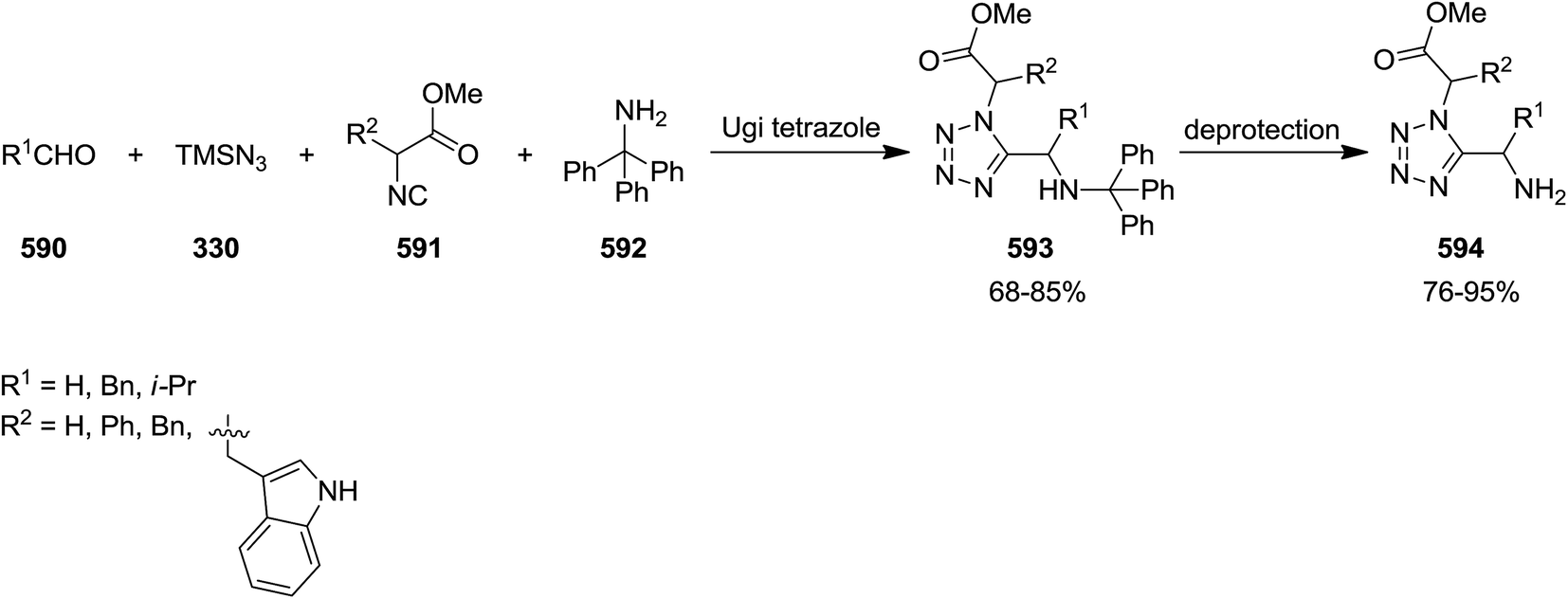
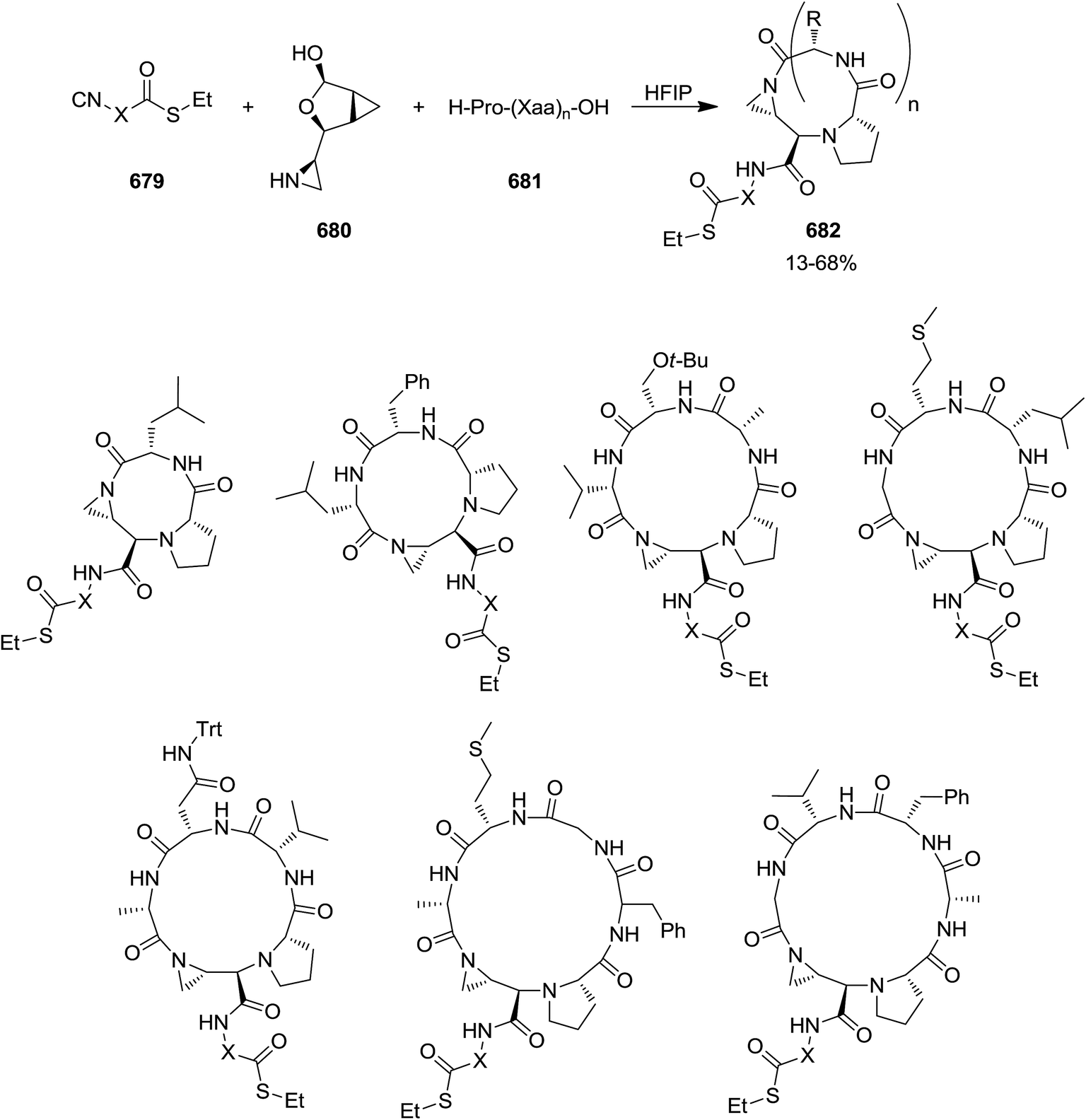
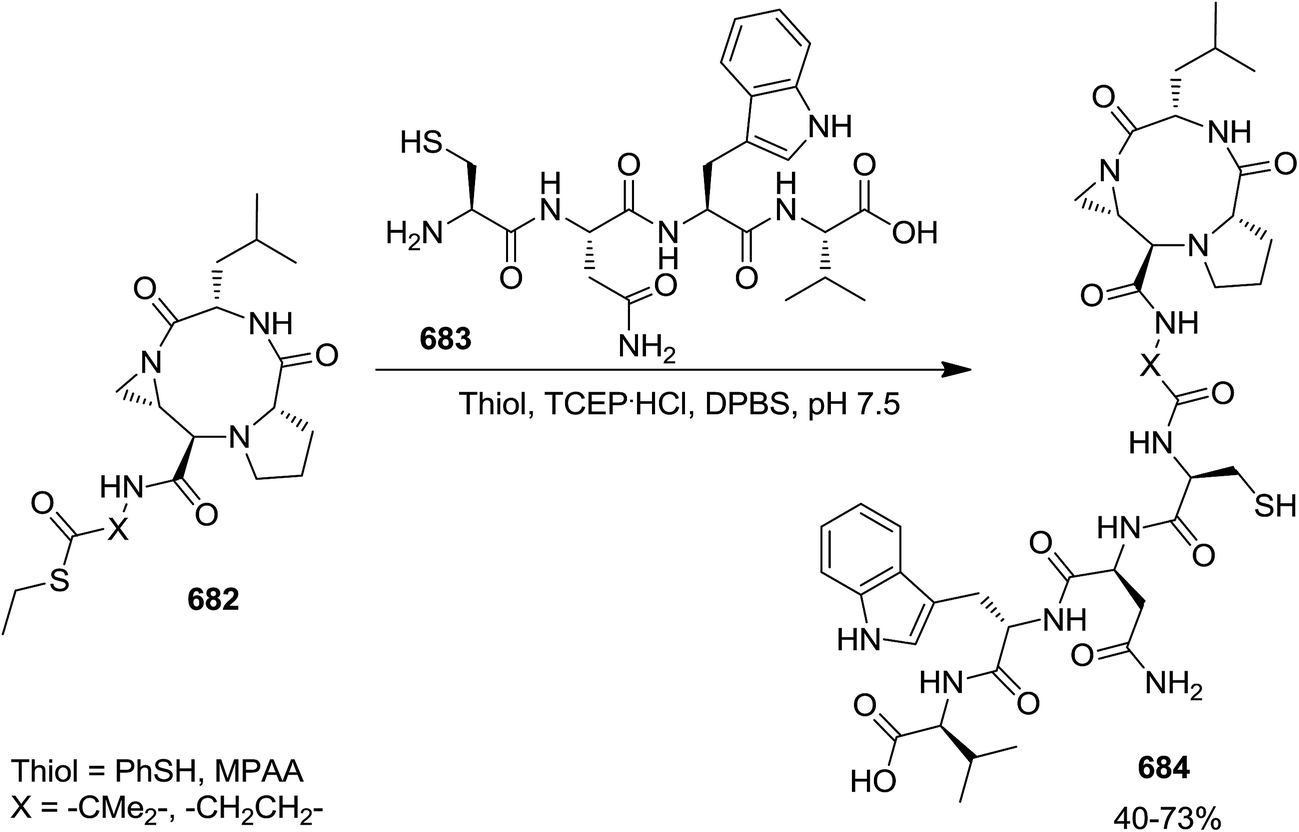
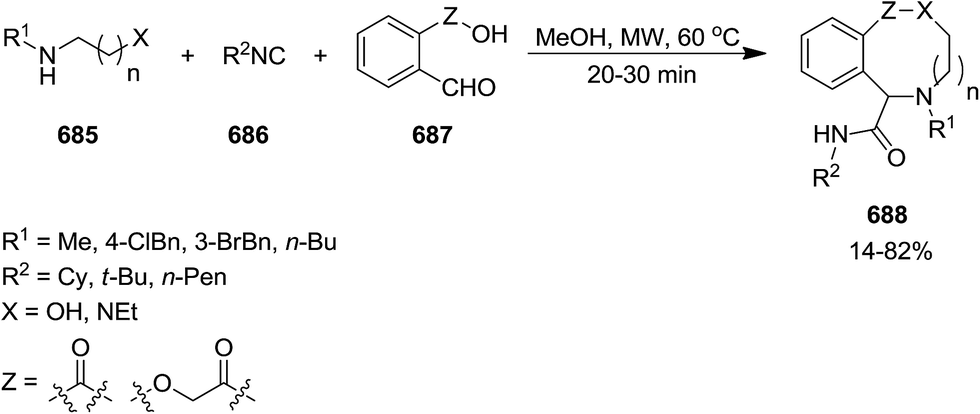
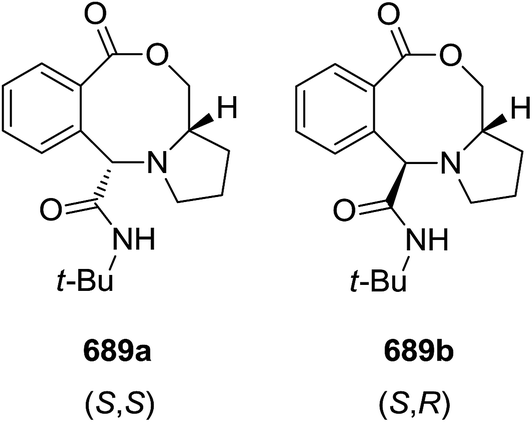
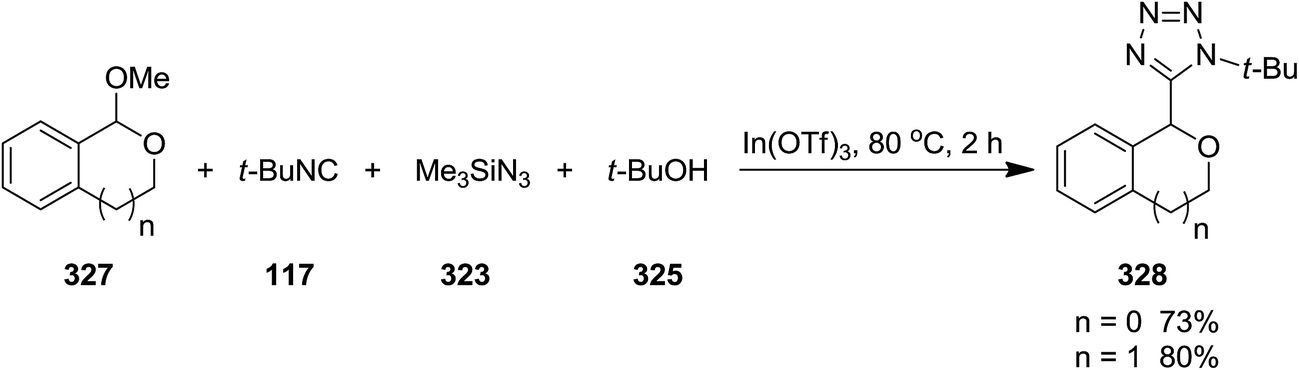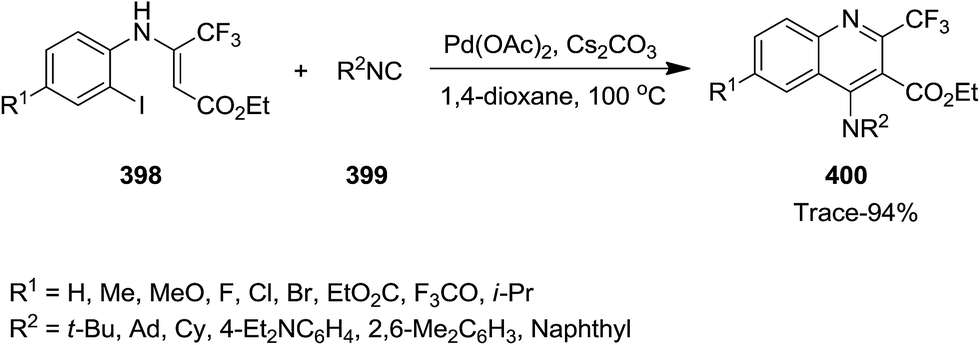DOI:
10.1039/C6RA02143C
(Review Article)
RSC Adv., 2016,
6, 53203-53272
Isocyanide-based multicomponent reactions in the synthesis of heterocycles
Received
24th January 2016
, Accepted 14th May 2016
First published on 17th May 2016
Abstract
In this review, we update our previous presentation, underscoring the recent applications of isocyanides as privileged synthons in the synthesis of various heterocyclic compounds, especially focused on those synthesized via multicomponent reactions.
 Samahe Sadjadi | Samahe Sadjadi was born in 1981 in Tehran, Iran. She received her PhD degree in 2009 after doing research in developing novel catalysts in organic chemistry at Alzahra University. She worked as Assessment Senior Expert of Nanotechnology and Green Energy in Hitech Technology and industry center and subsequently appointed as an assistant professor at Iran Polymer and Petrochemical Institute. She has co-authored several research papers and review articles in refereed journals and edited some books on nanomaterials including Nanoreactors. Her research interest includes catalysis, nanomaterials, and green chemistry. |
 Majid M. Heravi | Majid M. Heravi was born in 1952 in Mashhad, Iran. He received his B.Sc. degree from the National University of Iran in 1975 and his M.Sc. and Ph.D. degrees from Salford University, England in 1977 and 1980. He completed his doctoral thesis under supervision of late Jim Clarck in Salford University, England. He started his career as a research fellow in Daroupakhsh (a pharmaceutical company) in 1981 Tehran, Iran and joined as an assistant professor to Ferdowsi University of Mashhad, Iran in 1983 and promoted to associate professor in 1993 and full professor in 1997 in the aforementioned university. In 1999 he moved to Alzahra University of Tehran, Iran as professor of chemistry where he is still working in. He has previously been a visiting professor at UC Riverside, California, USA and Hamburg University, Hamburg, Germany. His research interests focus on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. |
 Niousha Nazari | Niosusha Nazari was born in 1987, Gorgan, Iran. She received her B.Sc. degree in Applied Chemistry from Ferdowsi University of Mashhad, Iran, in 2010 and her M.Sc. degree in Organic Chemistry from Alzahra University of Tehran, Iran, under the supervision of professor Majid. M. Heravi in 2014. Her research interests involve organic methodology catalysis, and application of multi-component reactions in the synthesis of new heterocyclic compounds. |
1. Introduction
Isocyanides (also called isonitriles or carbylamines) are organic compounds bearing the functional group –N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C.1 They are the isomers of the respective cyanides (–CN), hence they are designated by prefix iso. The organic segment is connected to the isocyanide group via the nitrogen atom, not via the carbon. They are used as privileged synthons for the synthesis of other organic compounds2–22 in particular various heterocycles,23–37 frequently via multicomponent reactions (MCRs) namely isocyanide multicomponent reactions (IMCRs).25,38–49 Noticeably isocyanides are susceptible to polymerization.50
C.1 They are the isomers of the respective cyanides (–CN), hence they are designated by prefix iso. The organic segment is connected to the isocyanide group via the nitrogen atom, not via the carbon. They are used as privileged synthons for the synthesis of other organic compounds2–22 in particular various heterocycles,23–37 frequently via multicomponent reactions (MCRs) namely isocyanide multicomponent reactions (IMCRs).25,38–49 Noticeably isocyanides are susceptible to polymerization.50
As a matter of fact, the unique features of these compounds make them good candidates for being selected for the synthesis of divergent types of heterocycles.51–69 Furthermore, their compatibility with domino and cascade reactions, has paved the way for developing novel atom and bond economy synthetic strategies.70–72
Isocyanide is a functional group with outstanding reactivity including the ability to react with both electrophiles and nucleophiles, simultaneously. Isocyanide-based multicomponent reactions (IMCR) are powerful tools for the synthesis of a wide variety of organic compounds.73–85 Using this functional group, diverse one-pot, domino and tandem synthetic strategies have been extensively developed86–90 which benefit from high degree of atom and bond economy, regio-, chemo- and stereoselectivity, reliability, requiring relatively mild reaction conditions and simplicity of work up procedures.91,92 IMCRs can be achieved using catalysts or under catalyst-free conditions.49 Isocyanides have also been applied for the synthesis of organic compounds via reaction with ortho-palladated complexes.93–97
We are interested in heterocyclic chemistry98–105 and their synthesis via MCRs.106–117 We also are keen to update the applications of name reactions in the total synthesis of natural products118–128 and applications of different synthons, reagents and catalysts in organic synthesis.129–135
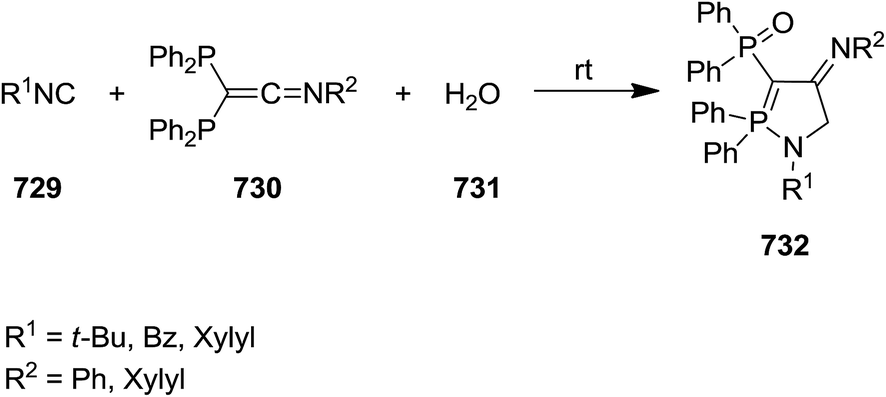
In 2011, we published a report entitled, Recent Applications of Isocyanides in the Synthesis of Heterocycles in Tetrahedron.36 Since then, a literature survey has revealed a plethora of publications in the use of isocyanides as privileged synthons in the synthesis of heterocycles, particularly those proceeding via multicomponent reactions. These heterocyclic compounds, including small ring, five, six and large membered rings as well as spiro heterocycles, often contain isocyanides as frameworks in their structures. In this review, we wish to update (2011–2015) our previous report. Given the influence of isocyanides in research in the synthesis of a wide variety of heterocyclic systems, we also tried to provide more insight into isocyanide chemistry, including some reaction mechanisms, and discuss their optimal reaction conditions.
2. Isocyanide-based synthesis of heterocycles
2.1. Small ring
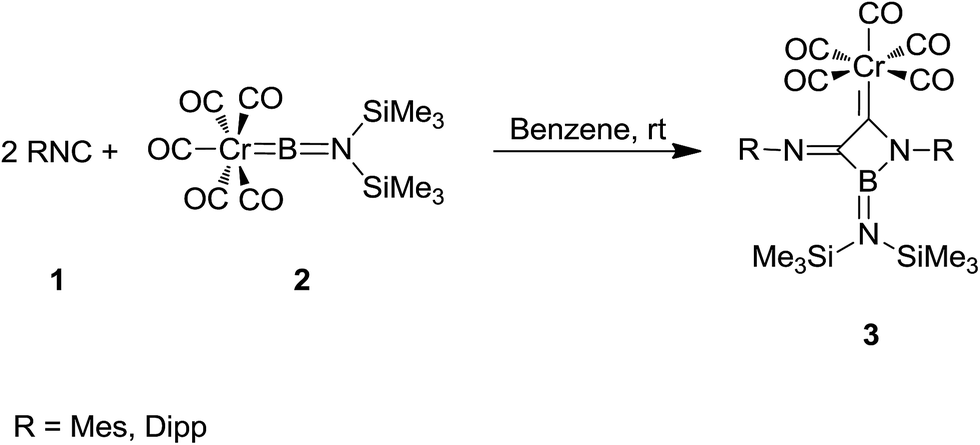
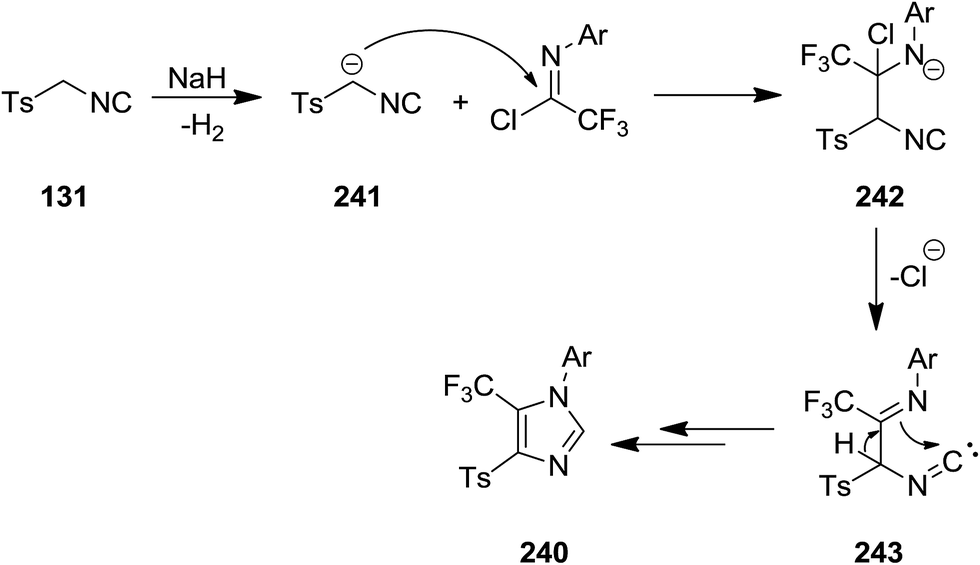
Braunschweig et al. reported the synthesis of boron-containing cyclic (amino)(imino)carbene complexes 3 from reaction of isocyanide 1 derived from MesNC (Mes = 2,4,6-Me3C6H2–) and DippNC (Dipp = 2,6-i-Pr2C6H2–) with chromium borylene complex [(OC)5Cr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BN(SiMe3)2] 2 (Scheme 1).136 The products were characterized by various techniques such as 11B NMR, 13C NMR, 1H NMR and single crystal X-ray diffraction. It was assumed that 3-membered BCN cyclic carbene complexes could be formed through [2 + 1] cycloaddition, followed by the isocyanide insertion.
BN(SiMe3)2] 2 (Scheme 1).136 The products were characterized by various techniques such as 11B NMR, 13C NMR, 1H NMR and single crystal X-ray diffraction. It was assumed that 3-membered BCN cyclic carbene complexes could be formed through [2 + 1] cycloaddition, followed by the isocyanide insertion.
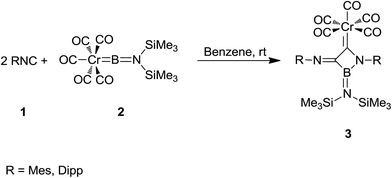 |
| | Scheme 1 | |
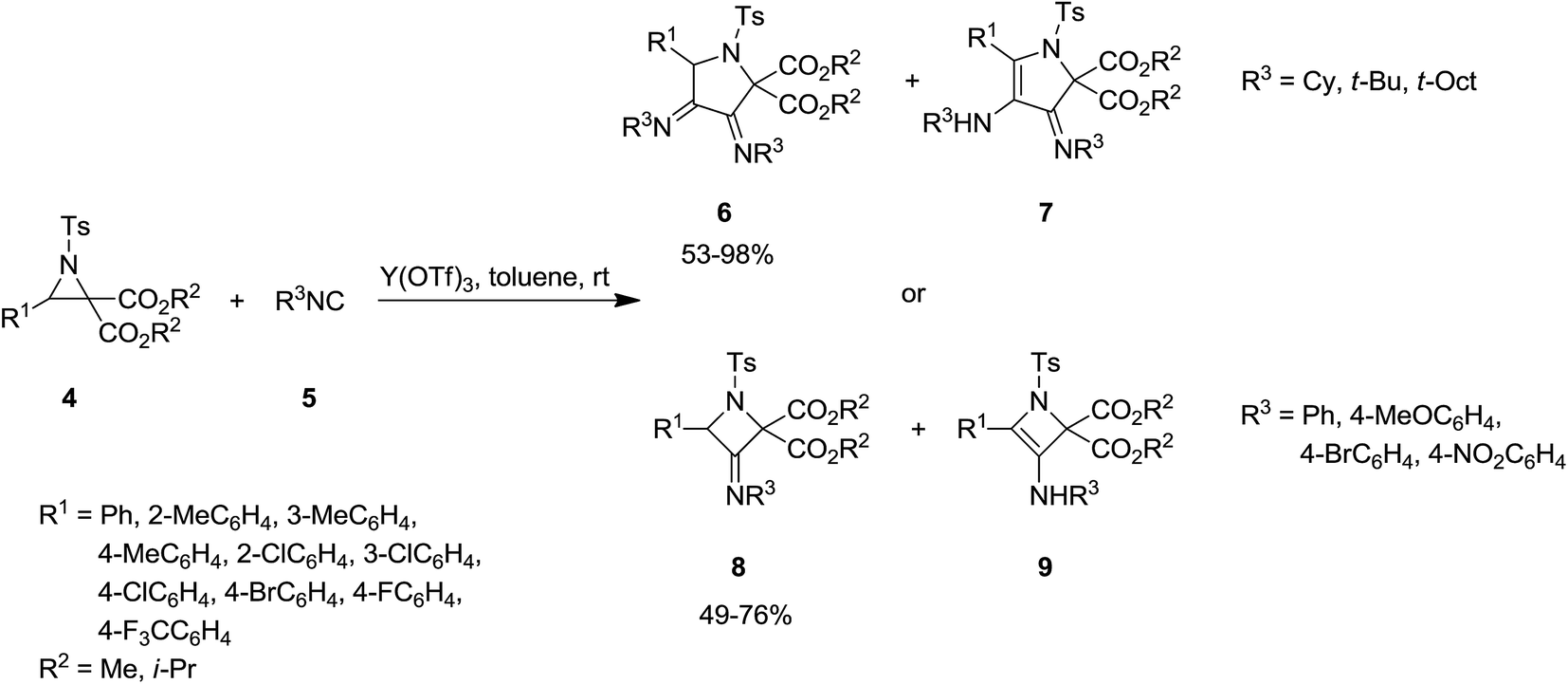
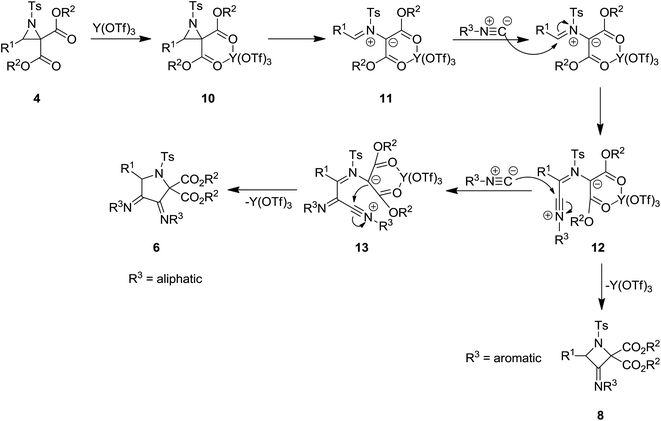
Y(OTf)3 was used as Lewis acid to catalyze [3 + 1 + 1] cycloaddition of N-tosylaziridine derivatives 4 and aliphatic isocyanides 5 at room temperature to afford the corresponding pyrrolidine 6 as major product as well as its isomer as minor product 7. Increasing the reaction time from 1 h to 20 h led to decrease of pyrrolidine yield and dominance of the formation of differing isomer (Scheme 2).137 Replacing aliphatic isocyanides with aromatic counterparts resulted in the formation of azetidine derivatives 8 and 9 via [3 + 1] annulations. The plausible mechanism is depicted in Scheme 3. Coordination of Lewis acid to dicarboxylate motif 10 followed by selective cleavage of C–C bond, produced an azomethine yield 11. Nitrilium intermediate 12 is generated by Ugi-type nucleophilic attack of the isocyanide. The latter was subjected to intermolecular addition with a second isocyanide molecule to afford 13. Next, intramolecular trapping of the nitrilium intermediate by the α-anion of the azomethine ylide occurred to afford the pyrrolidine derivative 6. In the case of azetidines, a similar mechanism may be proposed in which nitrilium intermediate could be trapped by α-anion.
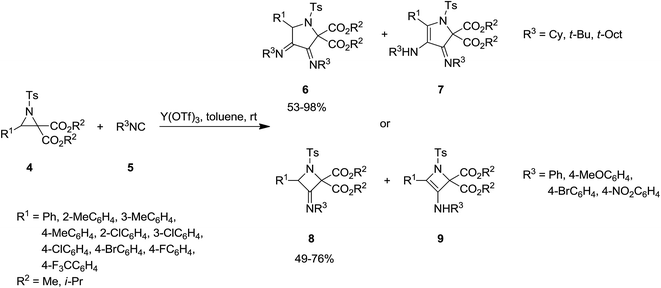 |
| | Scheme 2 | |
 |
| | Scheme 3 | |
2.2. Five-membered ring
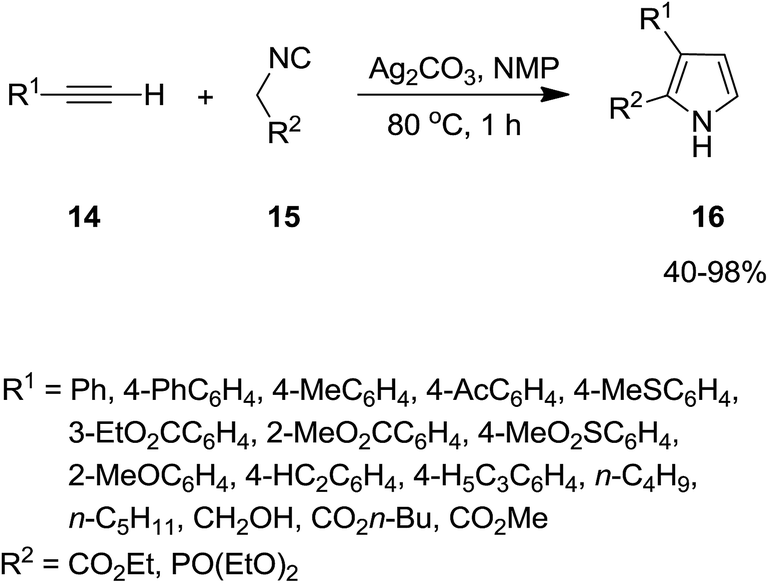
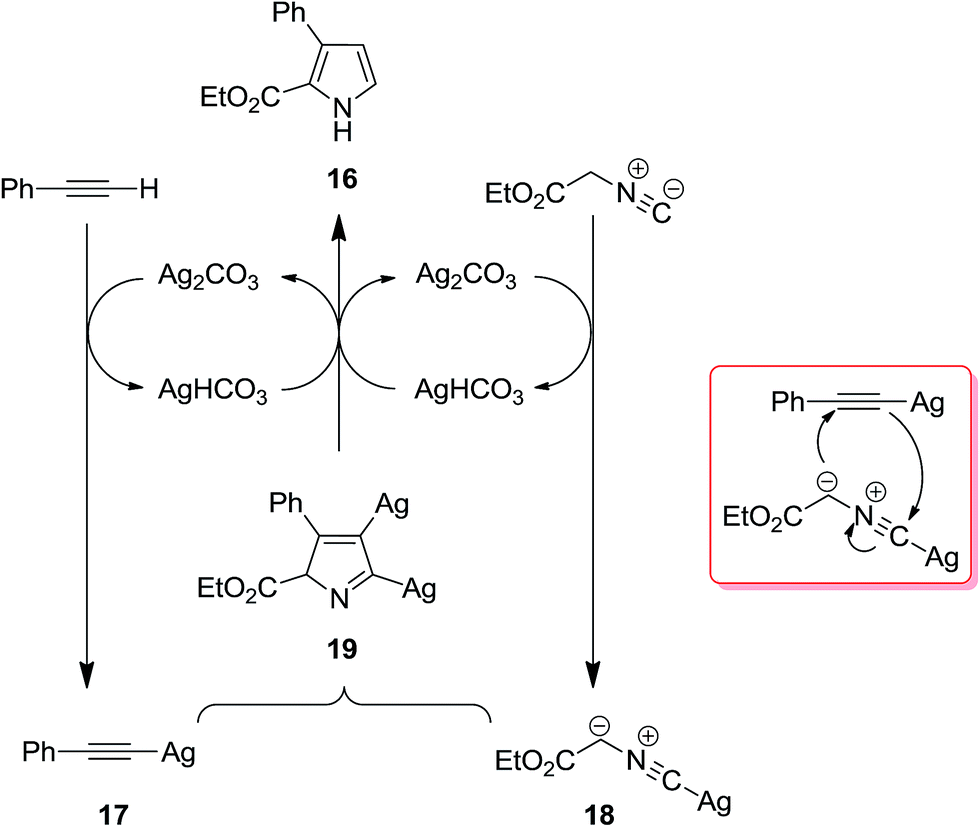
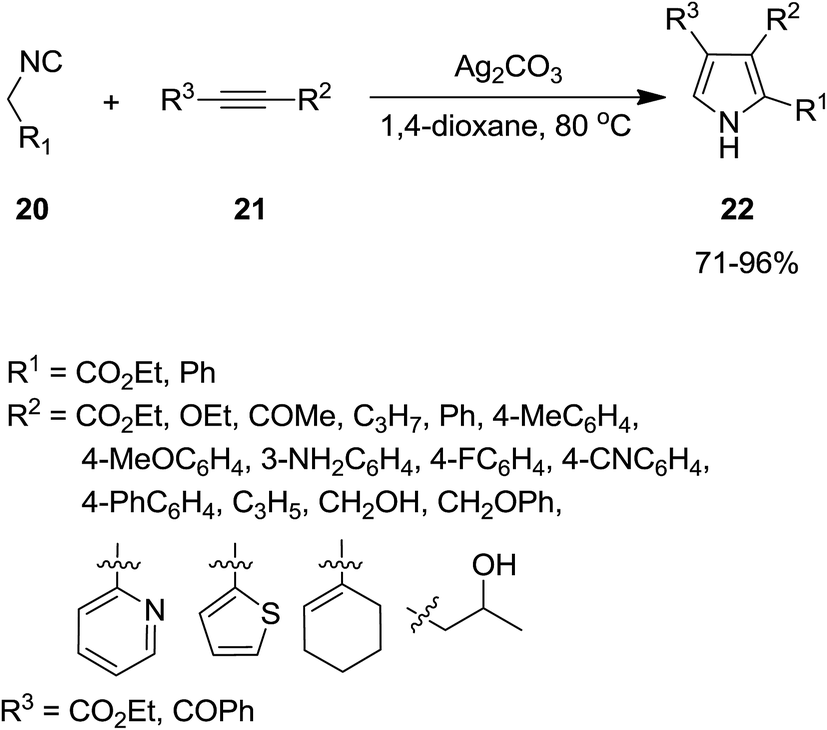
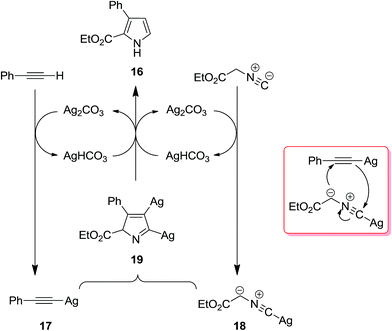
2.2.1 Pyrroles. Isocyanide-based syntheses of pyrrole derivatives have been studied extensively.138–145 Using Ag2CO3 as catalyst, Lei et al. developed an efficient, rapid and atom economy click-type synthetic method for preparing multi-substituted pyrroles 16. The reaction was based on co-cyclization of terminal alkynes 14 and isocyanide 15 (Scheme 4). The authors believed that reaction proceeded through the formation of silver-acetylide 17 and silver-isocyanide 18 complexes. The cycloaddition reaction of these two complexes afforded another intermediate 19 which underwent protonation and tautomerization to generate the corresponding pyrroles 16 (Scheme 5).146
 |
| | Scheme 4 | |
 |
| | Scheme 5 | |
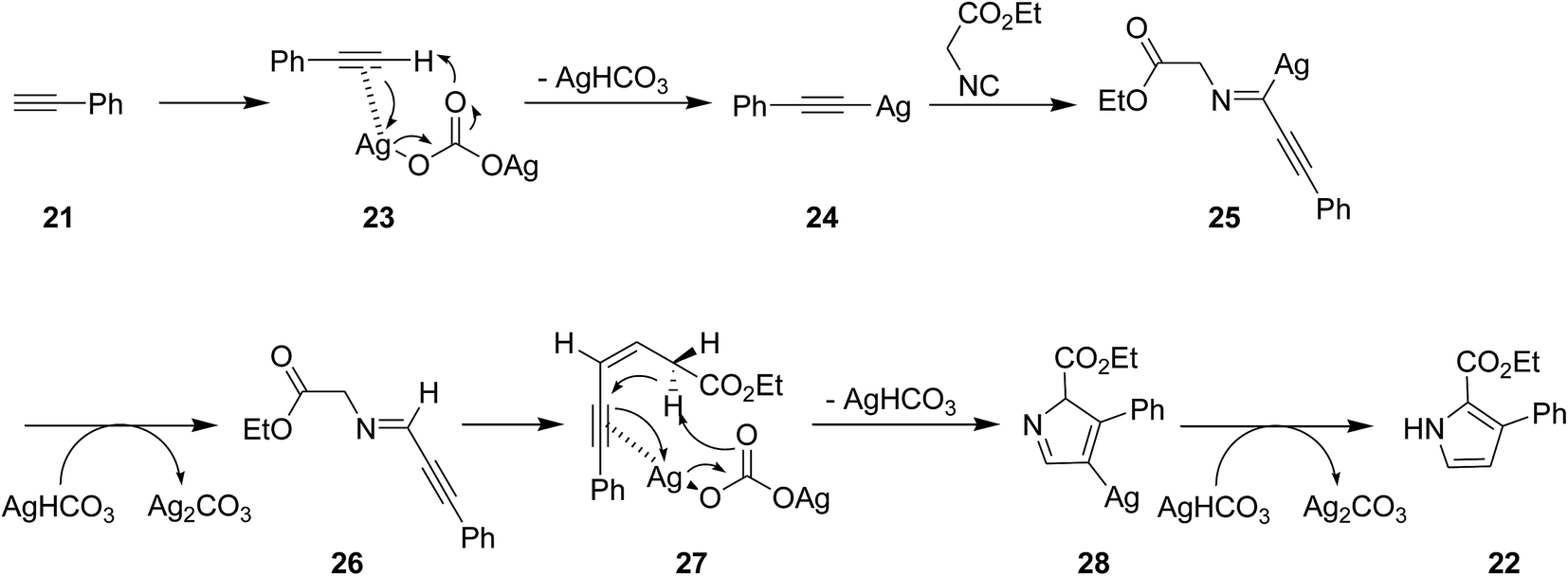
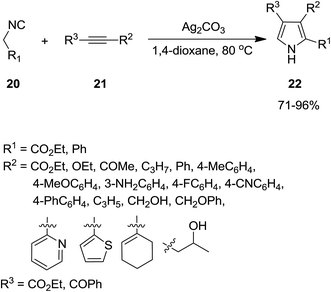
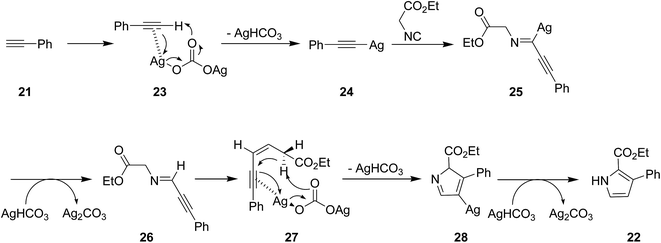
The utility of Ag2CO3 for catalyzing the cycloaddition of isocyanides 20 with alkyne derivatives 21 for efficient regioselective synthesis of 2,3-disubstituted and 2,3,4-trisubstituted pyrroles 22 was also proved (Scheme 6).147 The process exhibited broad range of substrate. The reaction mechanism was based on interaction of base 23 and acetylene 21 and formation of a silver-acetylide intermediate 24 followed by formation of an acetylenic imido complex 25 through 1,1 insertion of isocyanide into metal–carbon bond. Subsequently, photonolysis of the later with AgHCO3 occurred to afford acetylenic imide 26. The compound 28 could be obtained via interaction of Ag2CO3 and the intermediate 27, which subsequently tolerated 1,5-hydrogen shift and protonation to afford desired product (Scheme 7).
 |
| | Scheme 6 | |
 |
| | Scheme 7 | |
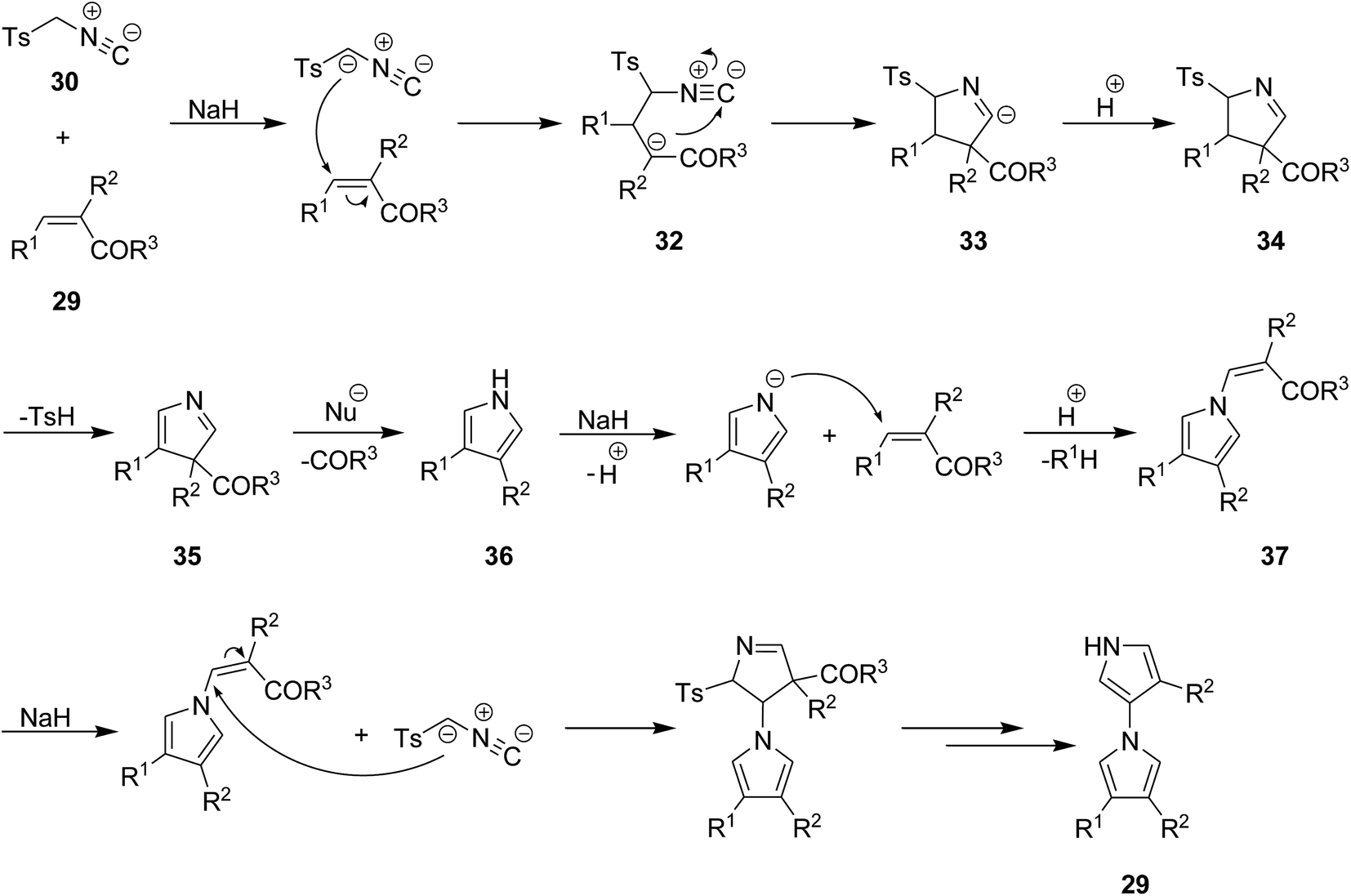
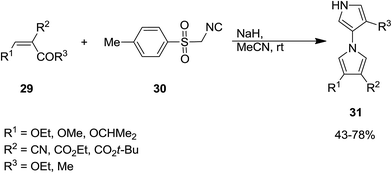
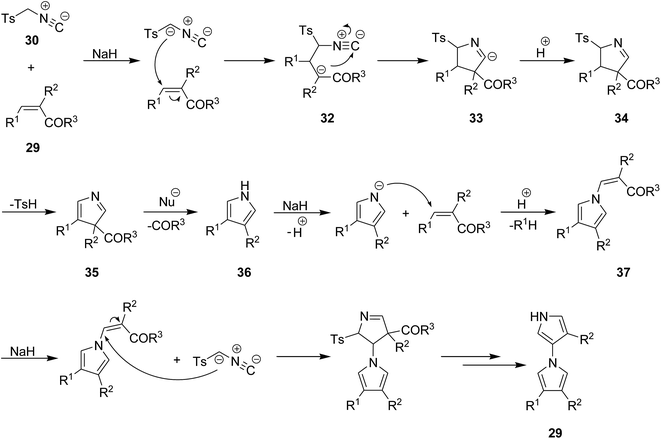
A mild and facile protocol was developed by Yu et al. for the synthesis of 1,3′-bipyrroles 31 in good yields. In this multi-component process poly-functionalized olefins 29 and tosylmethyl-isocyanide, TosMIC, 30 were used as starting materials. The reaction proceeded at room temperature in acetonitrile (Scheme 8).148 The proposed reaction mechanism involves, the formation of an intermediate 32 via reaction of TosMIC 30 with CC of olefin followed by intramolecular cycloaddition of isocyano moiety 33 and generation of a new intermediate 34. The latter then was transformed into a new intermediate 35 through removal of tosylsulphonic acid. This intermediate 35 was subjected to ester or ketone elimination to afford a new species 36 which reacted with the CC of second olefin to afford a new intermediate 37. The final product was furnished via repetition of the above-mentioned process (Scheme 9).
 |
| | Scheme 8 | |
 |
| | Scheme 9 | |
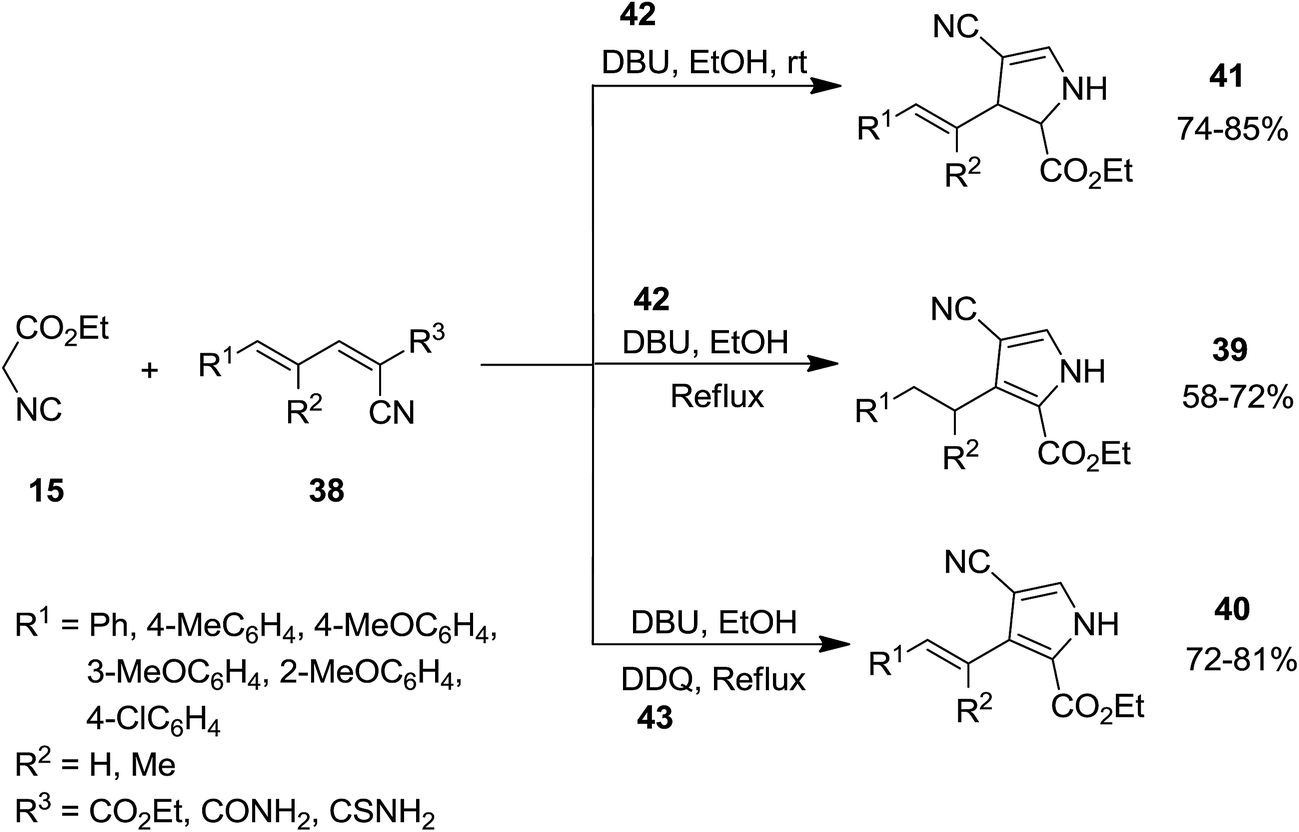
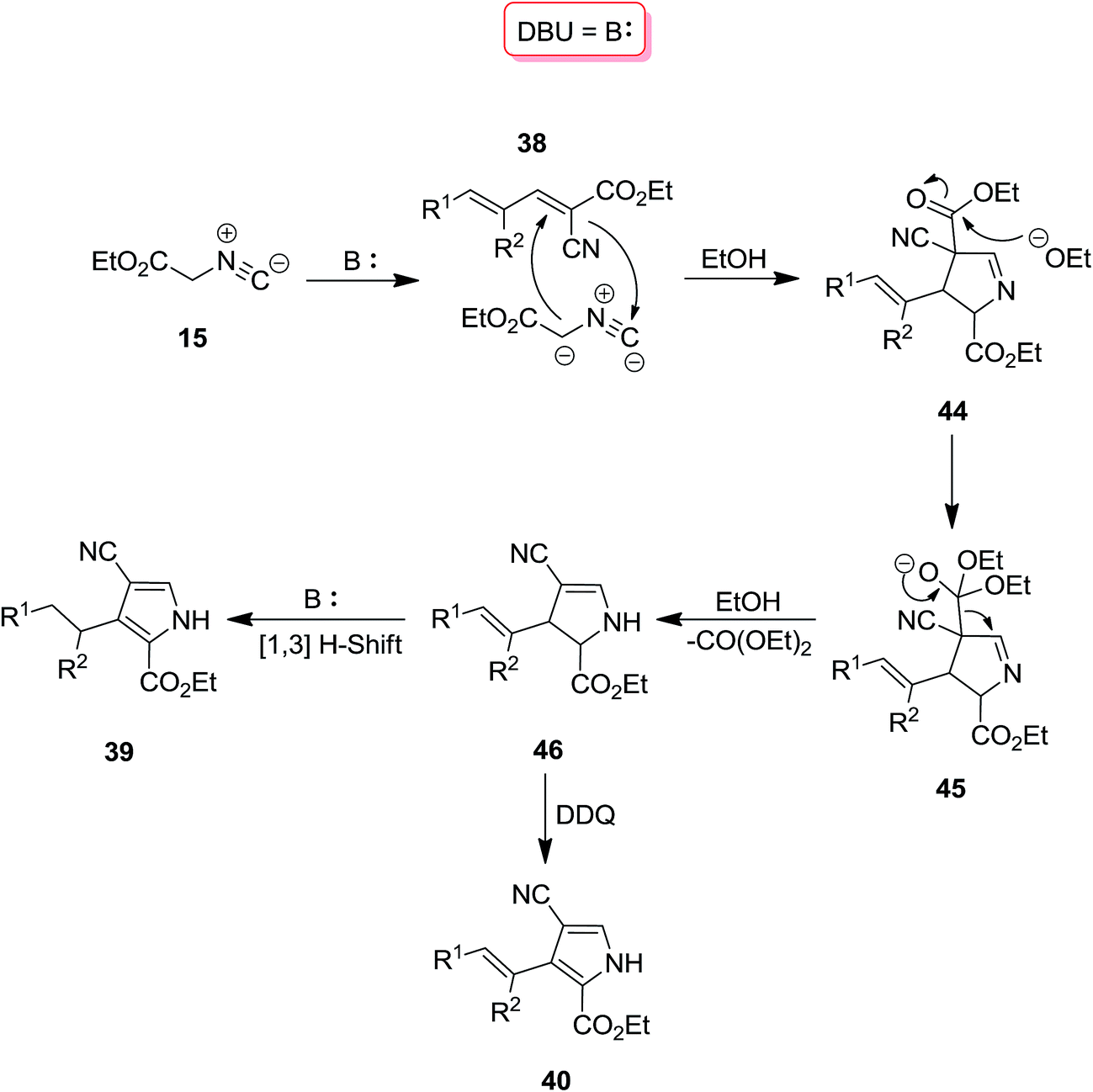
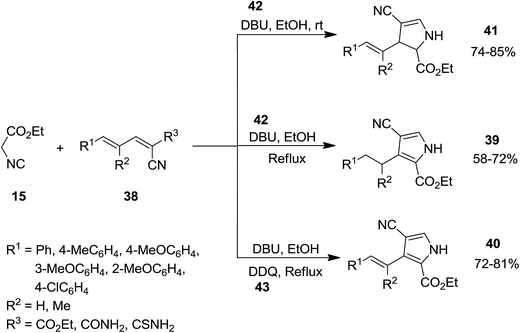
Using 2,4-pentadienenitriles 38 and ethyl isocyanoacetate 15 as starting materials, a novel one-pot three component protocol for preparation of biologically attractive pyrrole derivatives including, 2,3-dihydro-1H-pyrroles 39, 3-alkenyl-1H-pyrroles 40 and 3-alkyl-1H-pyrroles 41 has been developed. [2 + 3] cycloaddition of two reactants under mild reaction conditions, i.e. at room temperature in ethanol in the presence of 0.3 equivalent 1,8-diazabicyclo[5.4.0]undec-7-ene, DBU, 42 resulted in the formation of 2,3-dihydro-1H-pyrrole derivatives. Performing reaction under reflux condition and increasing DBU amount up to 2.0 equivalents led to the formation of 3-alkyl-1H-pyrroles. To obtain 3-alkenyl-1H-pyrroles, 1 equivalent of DDQ 43 was used as an oxidant under reflux condition (Scheme 10).149 The proposed reaction mechanism is depicted in Scheme 11. Deprotonation of 15 and its reaction with 38 resulted in the formation of compound 44. Intermediate 45 subsequently was transformed to 46 upon removal of diethyl carbonate.
 |
| | Scheme 10 | |
 |
| | Scheme 11 | |
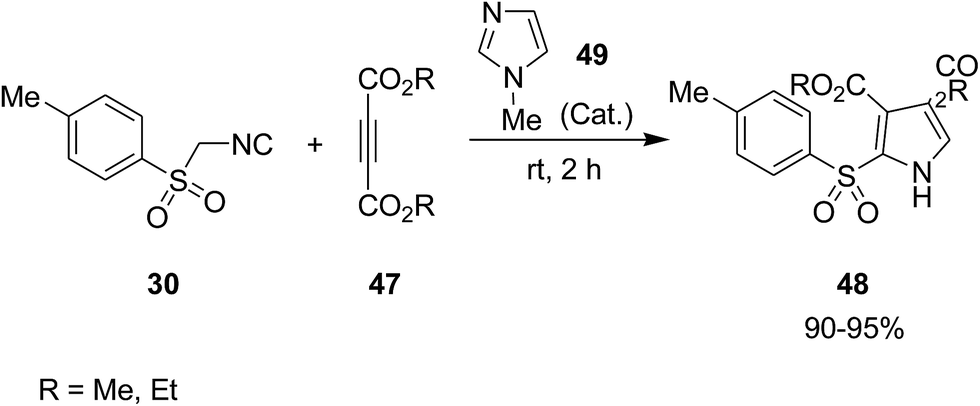
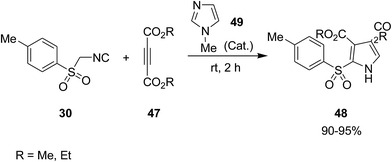
Poly-functionalized pyrroles 48, dialkyl 2-[(4-methylphenyl)sulfonyl]-1H-pyrrole-3,4-dicarboxylates, were synthesized via reaction of tosylmethyl isocyanide 30 and dialkyl acetylenedicarboxylates 47 under 1-methylimidazole 49 catalysis (Scheme 12).150 Simplicity, high yields, mild reaction conditions and high atom economy were the merits of this synthetic method.
 |
| | Scheme 12 | |
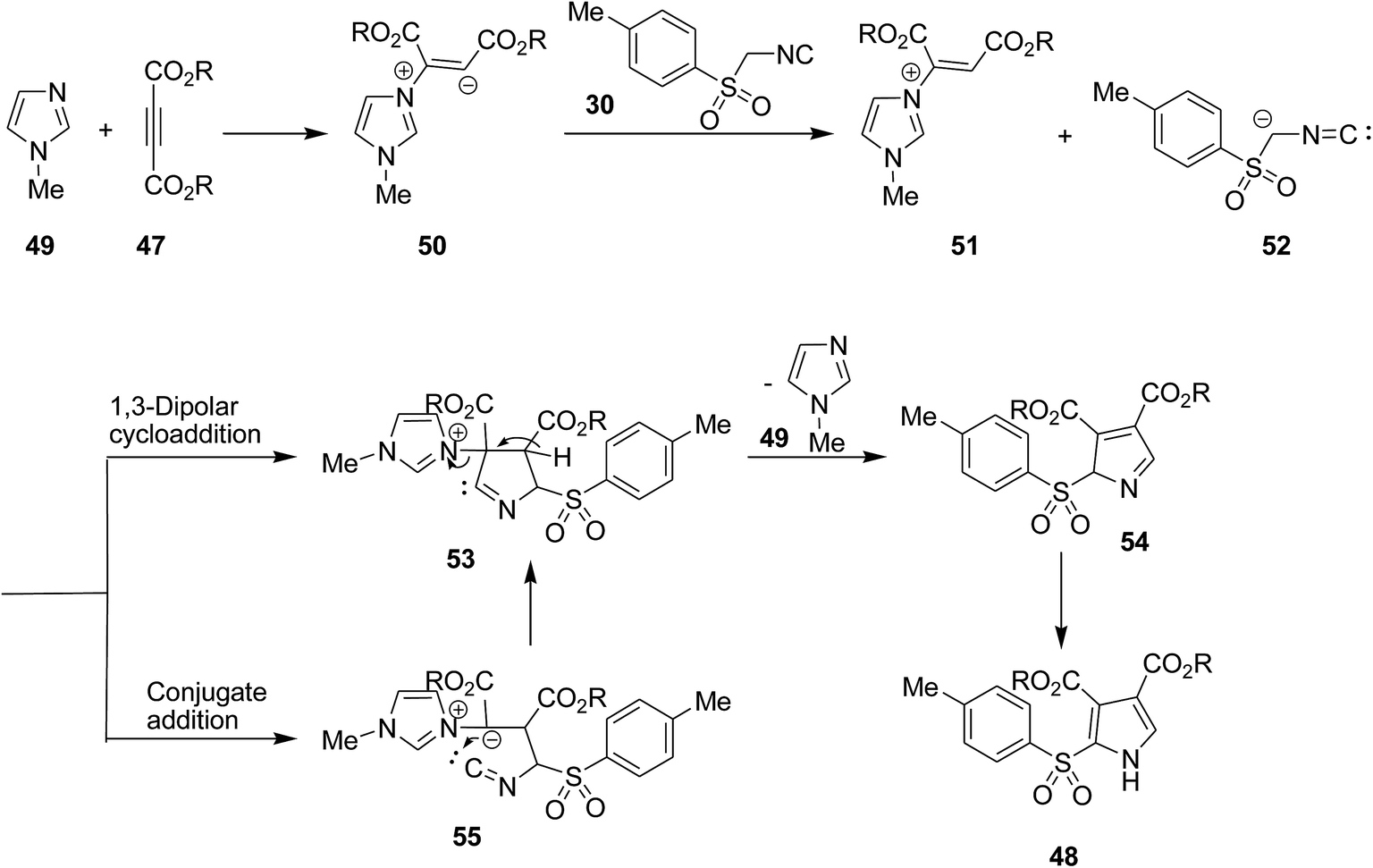
Mechanistically, zwitterionic intermediate 50 derived from addition of 1-methylimidazole 49 on the acetylenic ester 47 which is protonated to form 51. The latter is then reacted with anionic form of tosylmethyl isocyanide 52 which participated in 1,3-dipolar cycloaddition to afford a cyclic adduct 53. The latter tolerated 1-methylimidazole removal to furnish 54. This cyclic adduct could alternatively be generated via conjugate addition of anionic form of tosylmethyl isocyanide to protonated form of zwitterionic intermediate 55 which upon annulation along with the removal of catalyst led to the formation of a new intermediate which tautomerize to the final product 48 (Scheme 13).
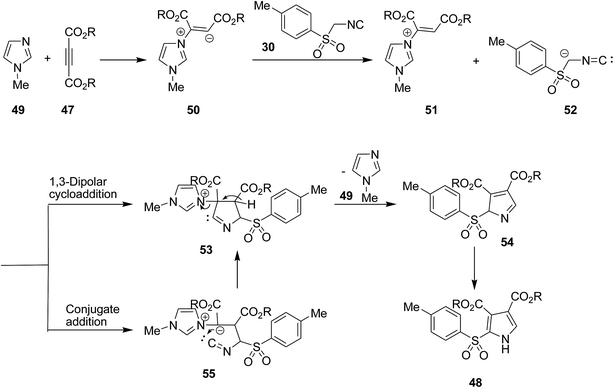 |
| | Scheme 13 | |
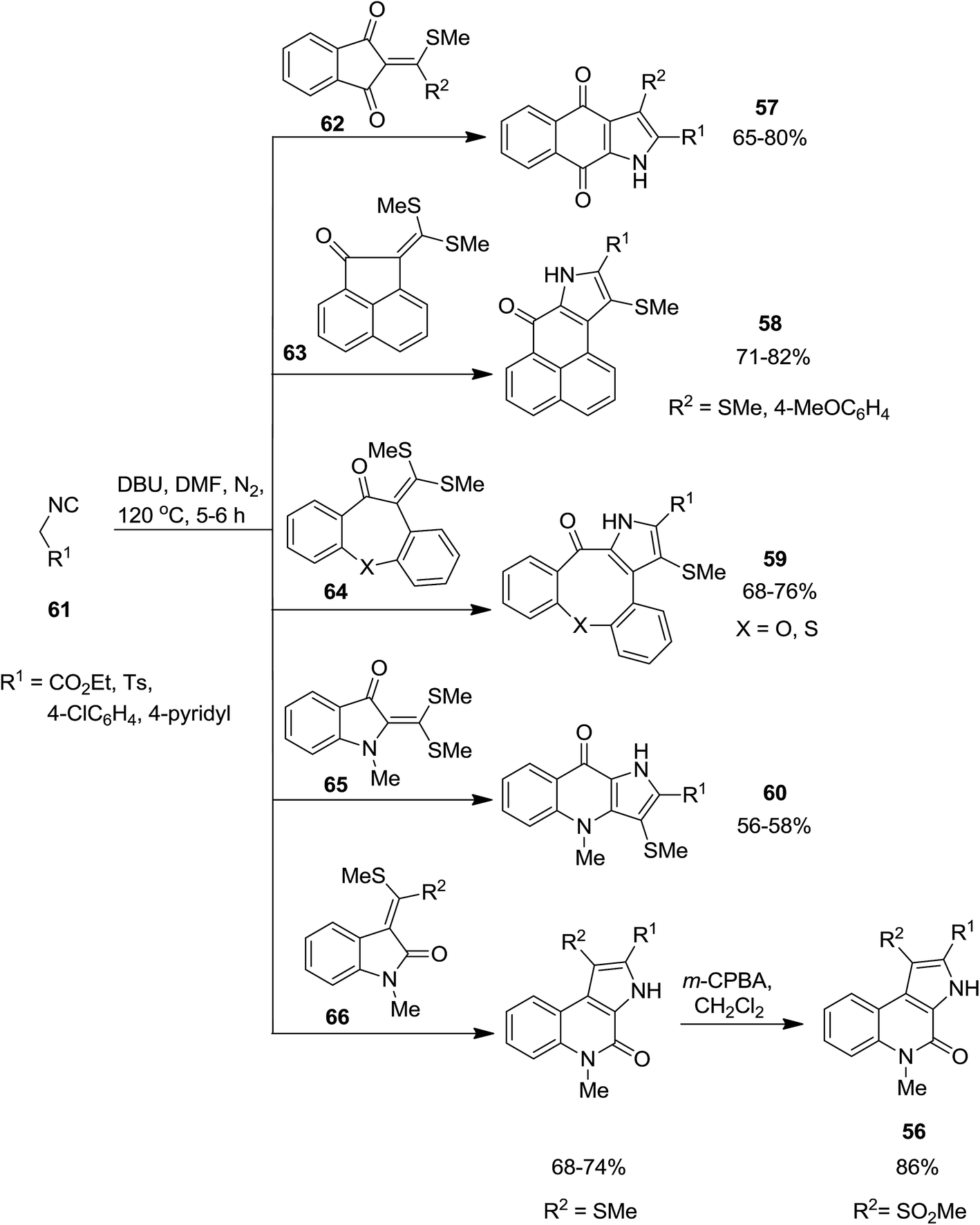
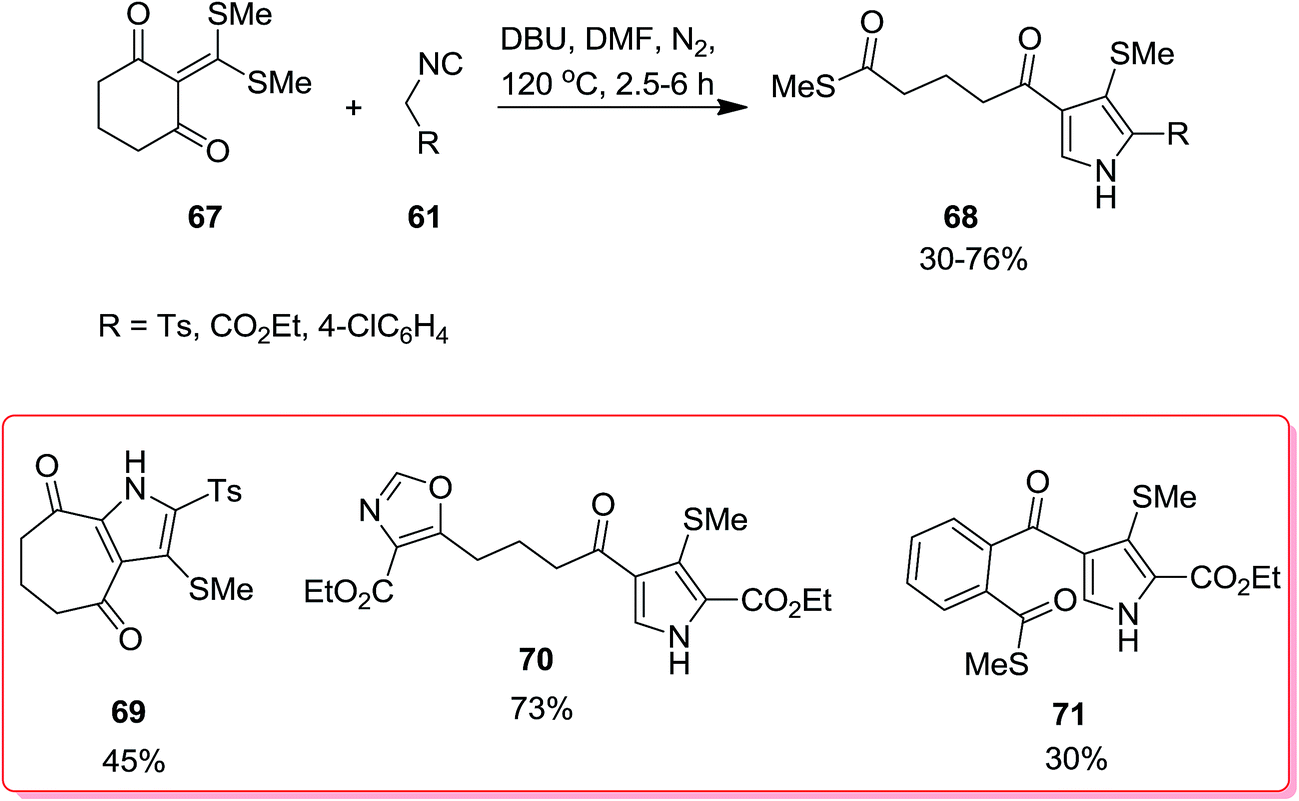
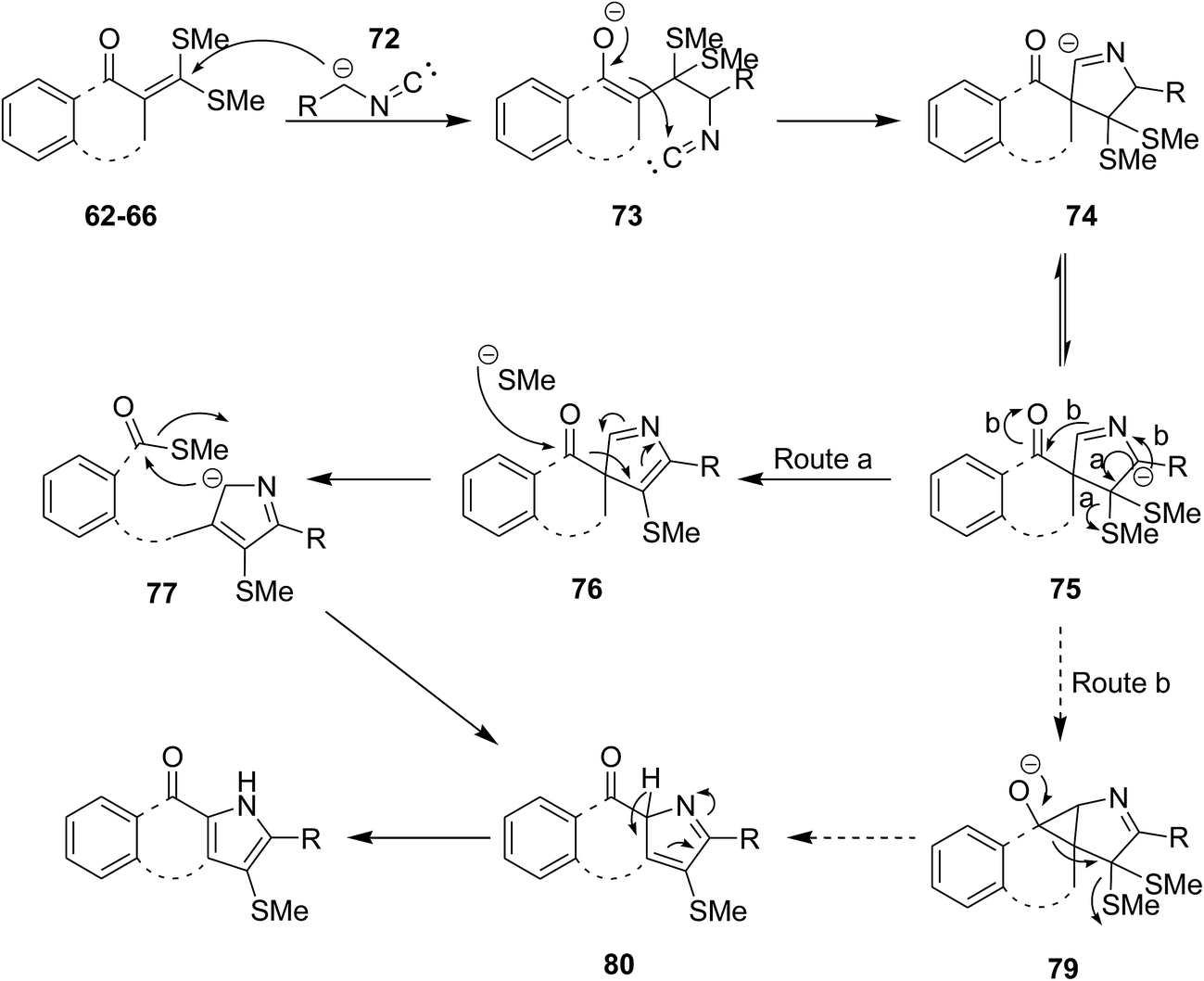
Biologically attractive fused pyrroles including tetracyclic fused indoles 56, angular and linear pyrroloquinolones 57, pyrrolonapthoquinones 58, pyrrole annulated dibenzooxocinone 59 and dibenzothiocinone derivatives 60 were synthesized via an innovative strategy. In this domino protocol, activated methylene isocyanides 61 reacted with cyclic α-oxoketene dithioacetals 62–66 under basic conditions (DBU was used as base) to provide annulated pyrroles (Scheme 14). Interestingly, cyclic ketones underwent one-carbon ring expansion in high regioselective manner. In the case of reaction of compound 67 with 61, different behavior was observed and products 68–71 were formed (Scheme 15). The authors proposed a mechanism in which oxoketene dithioacetals (62–66) were subjected to methylene isocyanide carbanion 72 and tolerated 1,4-conjugate addition to afford 73. Subsequently, intramolecular annulations occurred to afford unstable spiropyrrolenine anion 74 which was in equation to more stable aza-allyl anion 75. The later underwent methylthiolate anion removal to furnish spiroketone intermediate 76 (route a) followed by ring cleavage to give a new intermediate, ortho-substituted carbothioate, 77 which transformed into the final product via intramolecular cyclization along with elimination of methylthiolate anion 78. An alternative plausible mechanism (route b) contemplated for the generation of a strained tricyclic alkoxide intermediate 79 through intramolecular nucleophilic addition to the carbonyl group of aza-allyl anion. The later was then subjected into ring expansion via the elimination of methylthiolate anion 80 followed by isomerization to furnish the desired product (Scheme 16).151
 |
| | Scheme 14 | |
 |
| | Scheme 15 | |
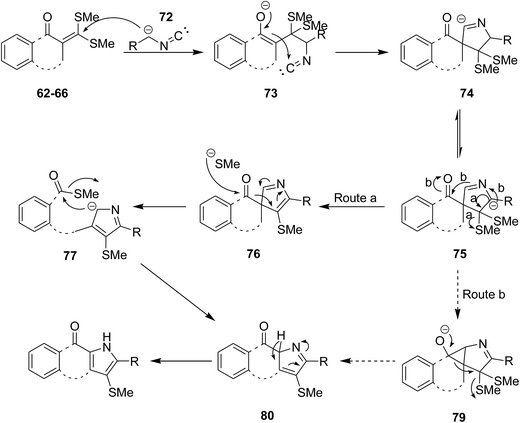 |
| | Scheme 16 | |
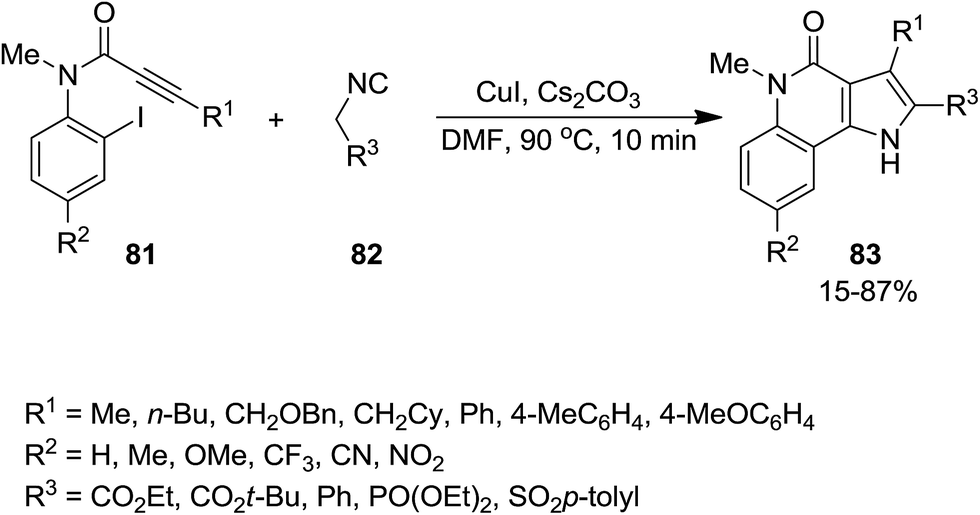
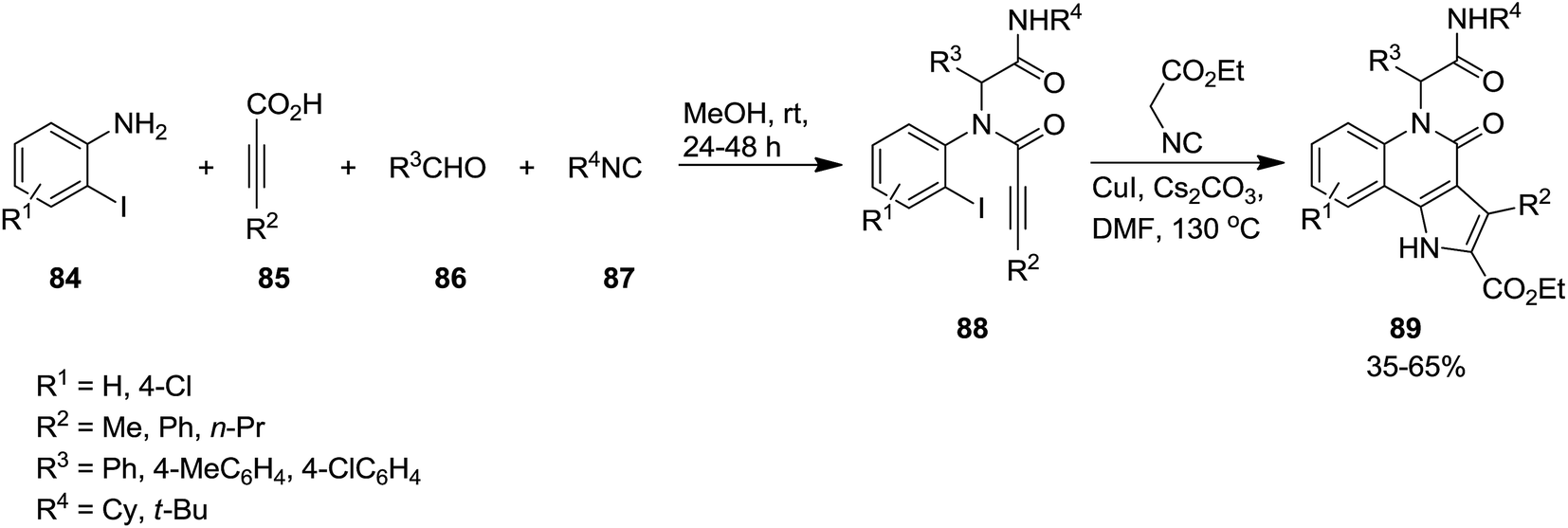
A novel synthetic approach based on cascade reaction of 1-(2-haloaryl)ynones 81 and isocyanides 82 under CuI catalysis provided biologically attractive 4-oxoindeno[1,2-b]pyrrole derivatives 83 (Scheme 17).152 Cs2CO3 and DMF were found to be the most effective base and solvent respectively. It is worth noting that the presence of a substituent at nitrogen was essential to obtain 4-oxoindeno[1,2-b]pyrroles in satisfactory yields since in the case of using N-unsubstituted pyrroles, just 4-protonated corresponding pyrroles were obtained as sole products. The authors also examined the two-step process consisting of Ugi four-component reaction of iodoaniline 84, acetylene derivatives 85, aldehydes 86 and isocyanide 87 for the formation of 88 followed by Cu-catalyzed cascade reaction to achieve more complicated derivatives 89 (Scheme 18).
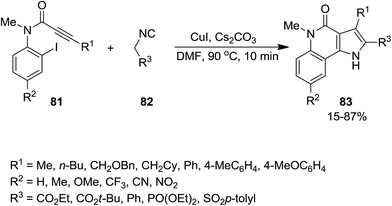 |
| | Scheme 17 | |
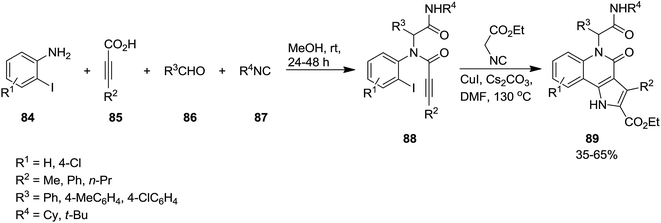 |
| | Scheme 18 | |
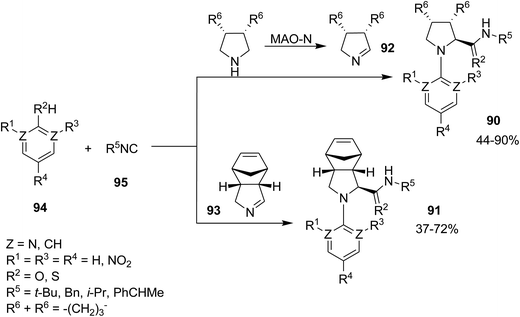
The first example of fully asymmetric Ugi–Smiles process was represented by Ruijter and Orru for the preparation of N-aryl prolineamide derivatives 90 and 91 in moderate to high yields but in excellent optical purity. This process involved asymmetric synthesis of substituted 1-pyrrolines 92 by using engineered monoamine oxidase N (MAO-N) or using pyrrolidine 93. The desired products were furnished by Ugi–Smiles reaction of isocyanides 94 and 95 (Scheme 19).153 The authors also expanded the scope of this synthetic protocol successfully to synthesize more complex Ugi–Smiles products containing reactive moieties such as allyl and azide groups which could participate in further reactions such as cycloaddition and olefin metathesis reactions.
 |
| | Scheme 19 | |
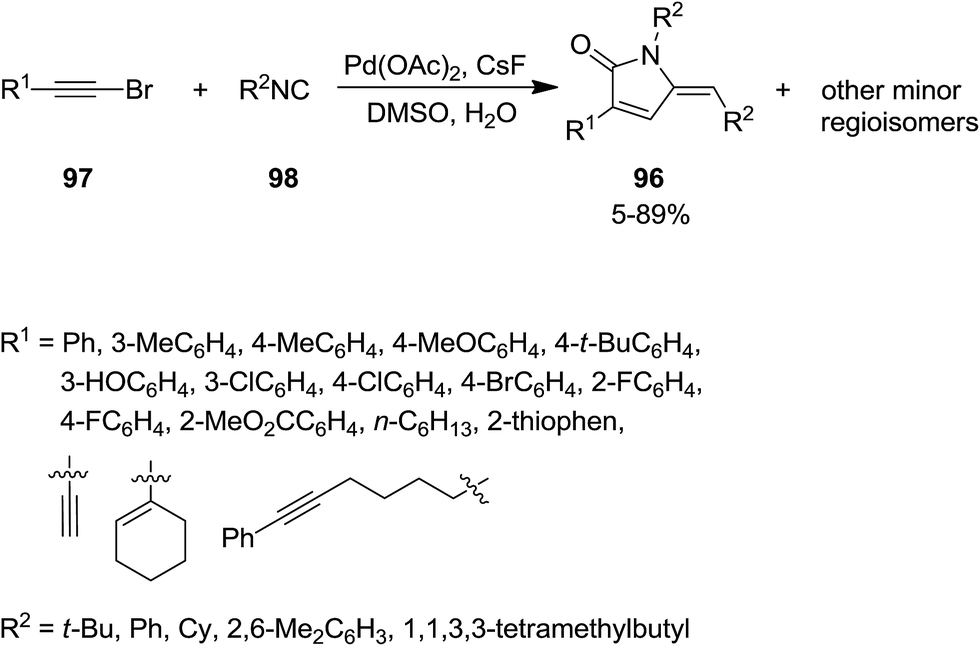
Using Pd as catalyst and CsF as co-catalyst, 5-iminopyrrolones 96 were obtained from a one-pot cyclization reaction of bromoalkynes 97 and isocyanides 98 in good yield and with high regioselectivity. This reaction showed broad substrate scope (Scheme 20).154 The plausible mechanism consisted of the formation of haloacrylamides as intermediates derived from the common nucleophilic addition of isocyanides to bromoalkynes.
 |
| | Scheme 20 | |
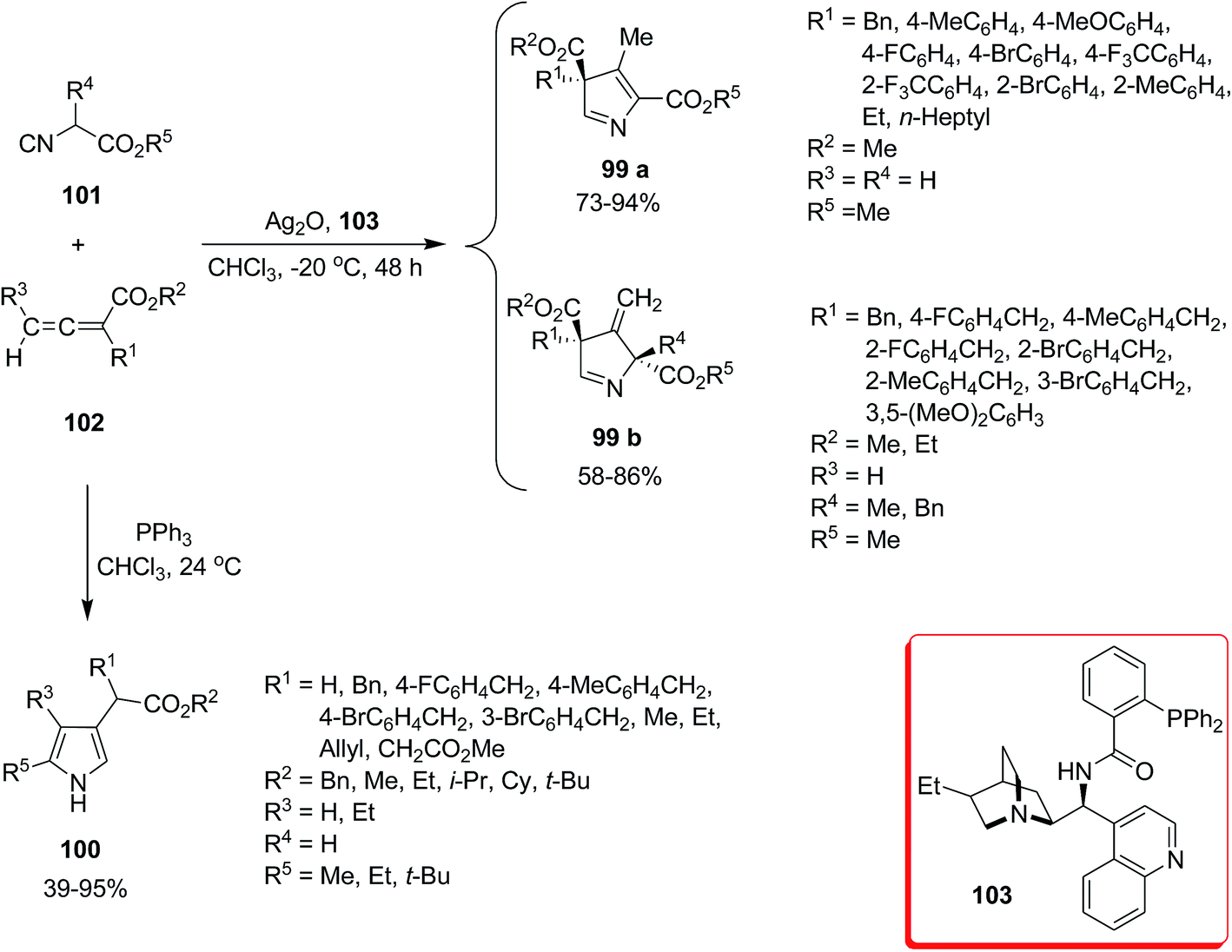
Zhao et al. developed a novel procedure for synthesis of 3H pyrroles 99a and 99b and 1H pyrrole derivatives 100 through [3 + 2] cyclization of activated isocyanides 101 and allenoates 102 under Ag2O or PPh3 catalysis.155 Using Ag catalyst, 3H pyrroles 99a and 99b were obtained enantioselectively (Scheme 21). In this procedure, the ligand 103 played an important role in accelerating and moderating the reaction condition and increasing the yields of products. The generality of reaction was proved by applying various allenoates and isocyanoacetates. The use of PPh3 as catalyst led to formation of 1H pyrrole derivatives 100 in high yields.
 |
| | Scheme 21 | |
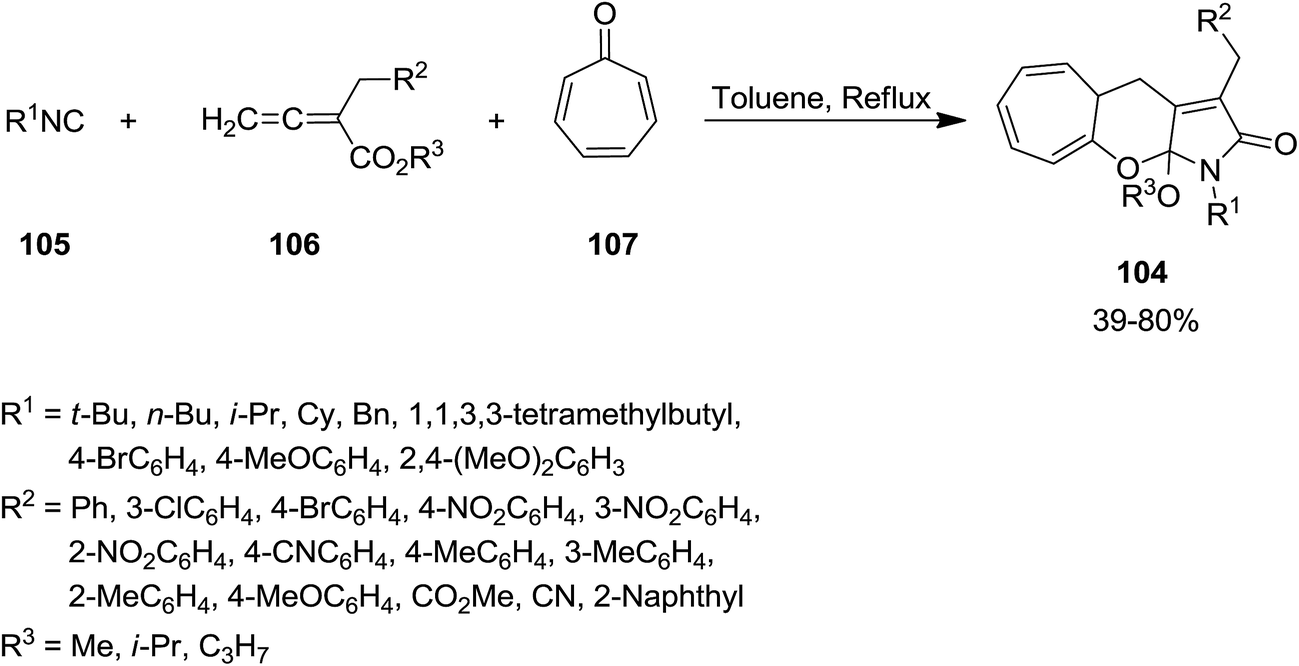
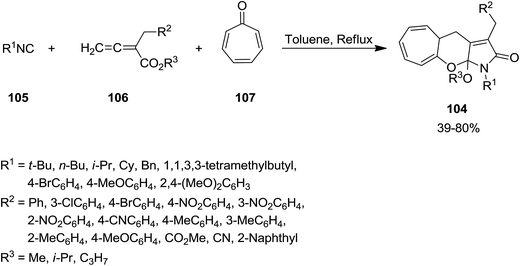
Li, Jia and coworkers reported efficient synthesis of a series of tricyclic heterocyclic framework 104 through three-component tandem cycloaddition between isocyanide 105, allenoate derivatives 106 and tropone 107 under reflux condition in toluene (Scheme 22).156 The protocol showed excellent substrate scope and allenoate derivatives with various electron-donating and electron-withdrawing groups could furnish the desired products. Moreover, both aromatic and aliphatic isocyanides could be used in this procedure. The proposed mechanism was based on [8 + 2 + 1] cycloaddition, [1,5]-H shift, and cyclization and alkoxy group migration. Good to high yields, good atom economy and mild reaction condition were the merits of the procedure.
 |
| | Scheme 22 | |
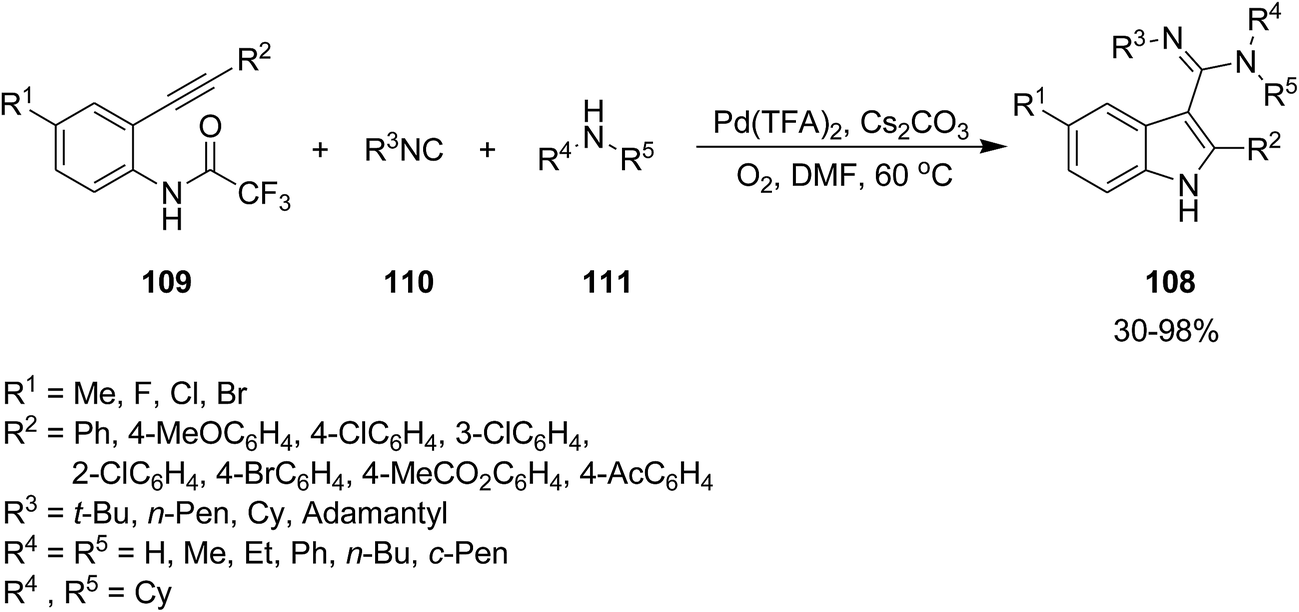
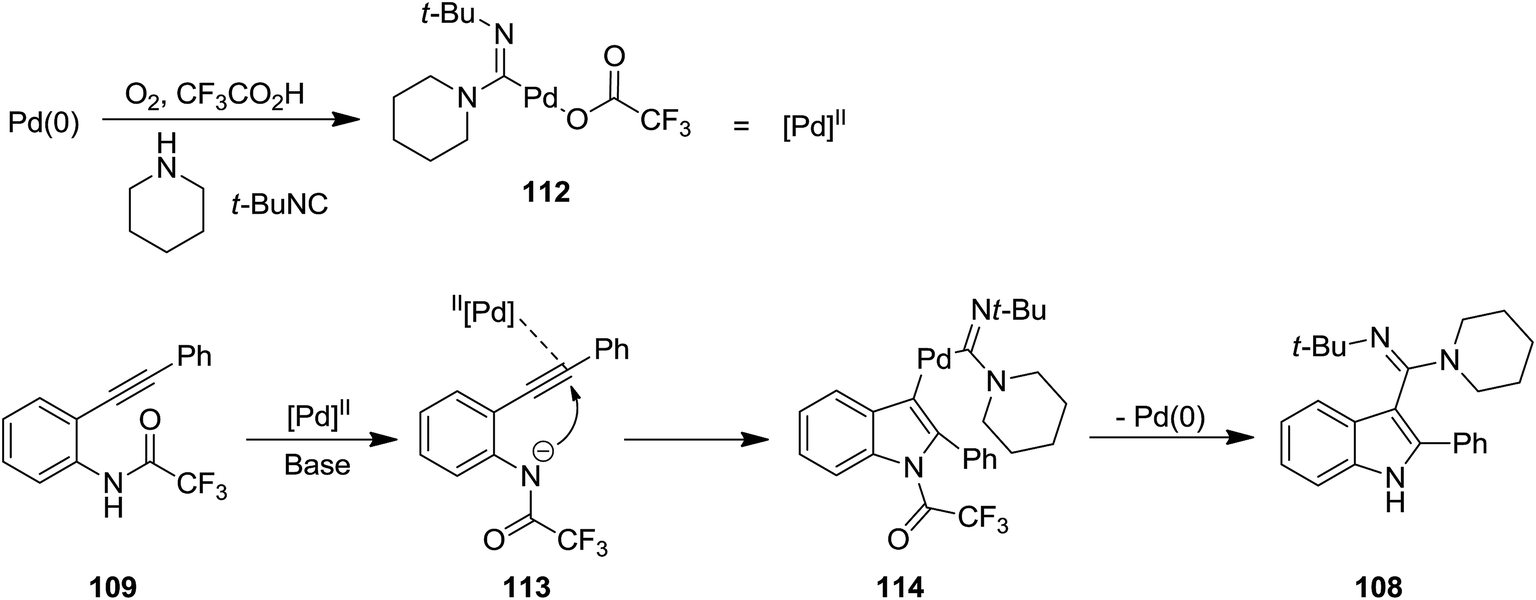
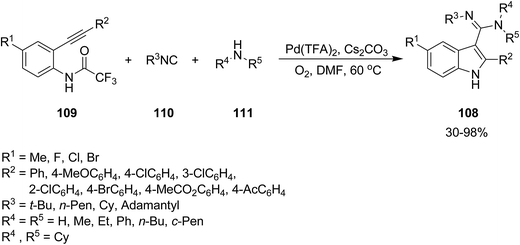
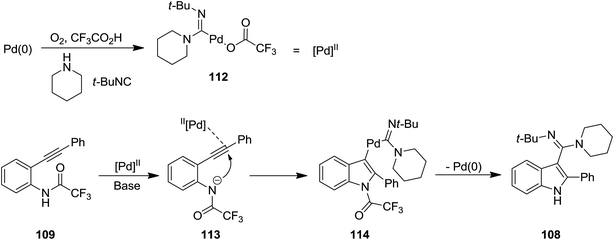
2.2.2 Indole. The attractive properties of indoles and their broad applications make them one of the most studied heterocycles. There are numerous reports in the literature regarding the utility of isocyanide chemistry for synthesis of various indole derivatives.157–170 2-Substituted 1H-indole-3-carboxamidine derivatives 108 were obtained in good yields via multi-component reaction of o-alkynyltrifluoroacetanilides 109, isocyanides 110 and amines 111 in the presence of Cs2CO3 as base, under Pd(II) catalysis (Scheme 23).171 It is worth mentioning that the presence of O2 is essential for progress of this reaction since it did not proceed under inert gas atmosphere. The proposed mechanism consisted of the formation of an active Pd(II) complex 112 in the presence of isocyanide and amine and its subsequent coordination to o-alkynyltrifluoroacetanilides 113. An intermediate 114 was generated via intramolecular attack of anilide anion. Finally, the reductive elimination and detrifluoroacetylation gives the desired product (Scheme 24).
 |
| | Scheme 23 | |
 |
| | Scheme 24 | |
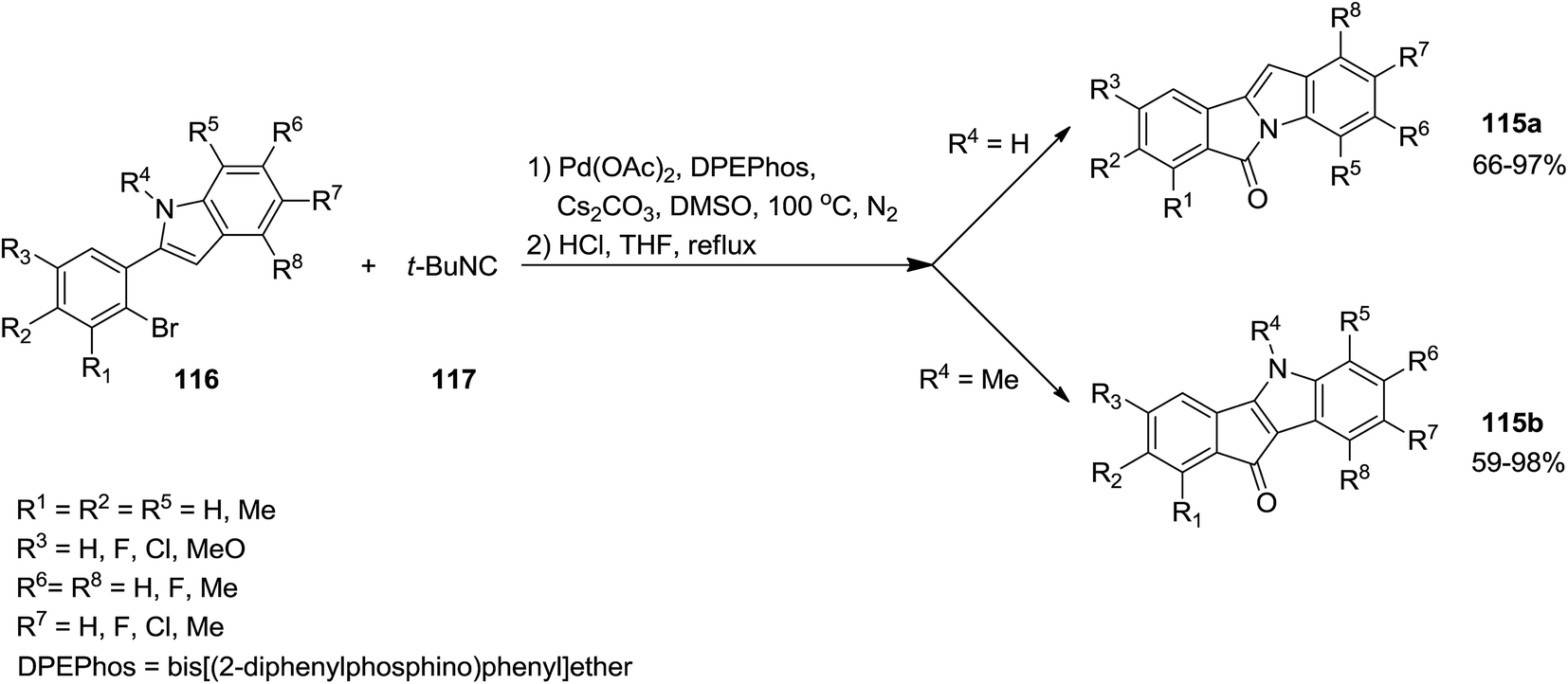
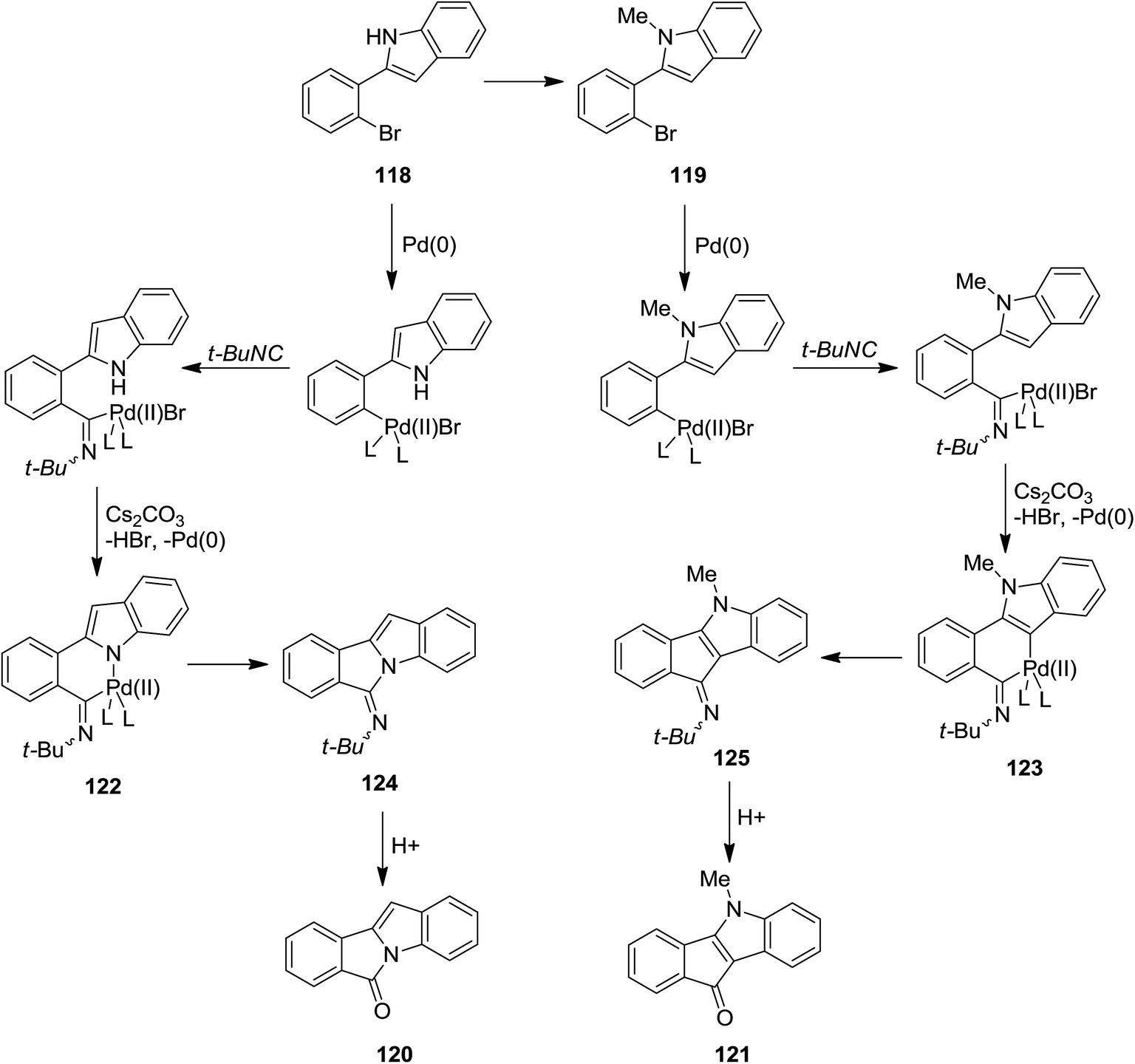
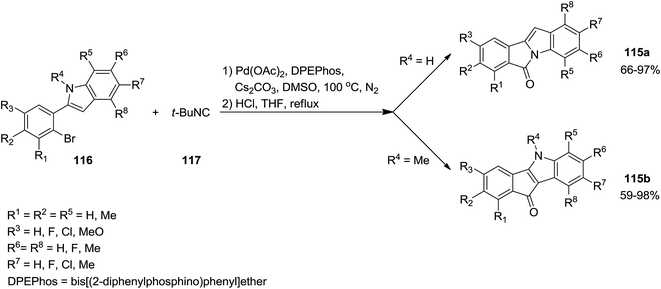
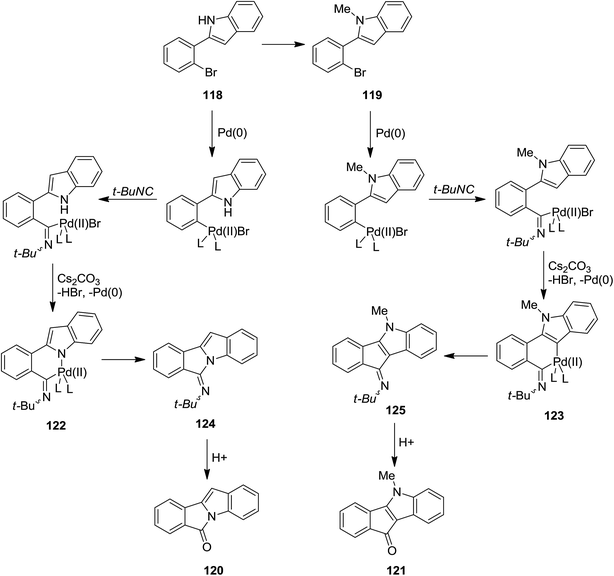
A novel synthetic method for the formation of biologically active 6H-isoindolo[2,1-a]indol-6-ones 115a and indenoindolone derivatives 115b was developed by Zhu et al.172 This process was based on tert-butyl isocyanide 117 insertion (Scheme 25). The Pd-catalyzed reaction of tert-butyl isocyanide with 2-(2-bromophenyl)-1H-indoles or N-methyl or N-Boc protected 2-(2-bromophenyl)-1H-indoles 116 afforded 6H-isoindolo[2,1-a]indol-6-ones and indenoindolones respectively. This study proved the importance of using isocyanides in the formation of C–C or C–N bonds. The proposed mechanism is depicted in Scheme 26. Evidently, tert-butyl isocyanide insertion, which led to the formation of compounds 120 and 121, occurs to a palladium complexes 118 and 119 which in turn were obtained from oxidative addition of 2-(2-bromophenyl)-1H-indole to palladium. The elimination of HBr led to the formation of intermediates 122 and 123 which were then subjected to reductive elimination to generate 124 and 125 followed by acid hydrolysis to afford the desired products.
 |
| | Scheme 25 | |
 |
| | Scheme 26 | |
Singh et al. reported synthesis of a series of bis(indolyl)methane derivatives 129 with antibacterial properties.173 Initially, 4-(di(1H-indol-3-yl)methyl)benzoic acid was obtained via reaction of p-carboxybenzaldehyde and indole in EtOH under mild reaction conditions. Subsequently, bis(indolyl)methane adducts 129 were obtained in high yields through Ugi and Passerini reactions of 4-(di(1H-indol-3-yl)methyl)benzoic acid 126, isocyanides 87, appropriate aromatic aldehydes 127 and aniline 128 (Scheme 27). The generality of reaction was established by using various isocyanides and differently substituted aromatic aldehydes.
 |
| | Scheme 27 | |
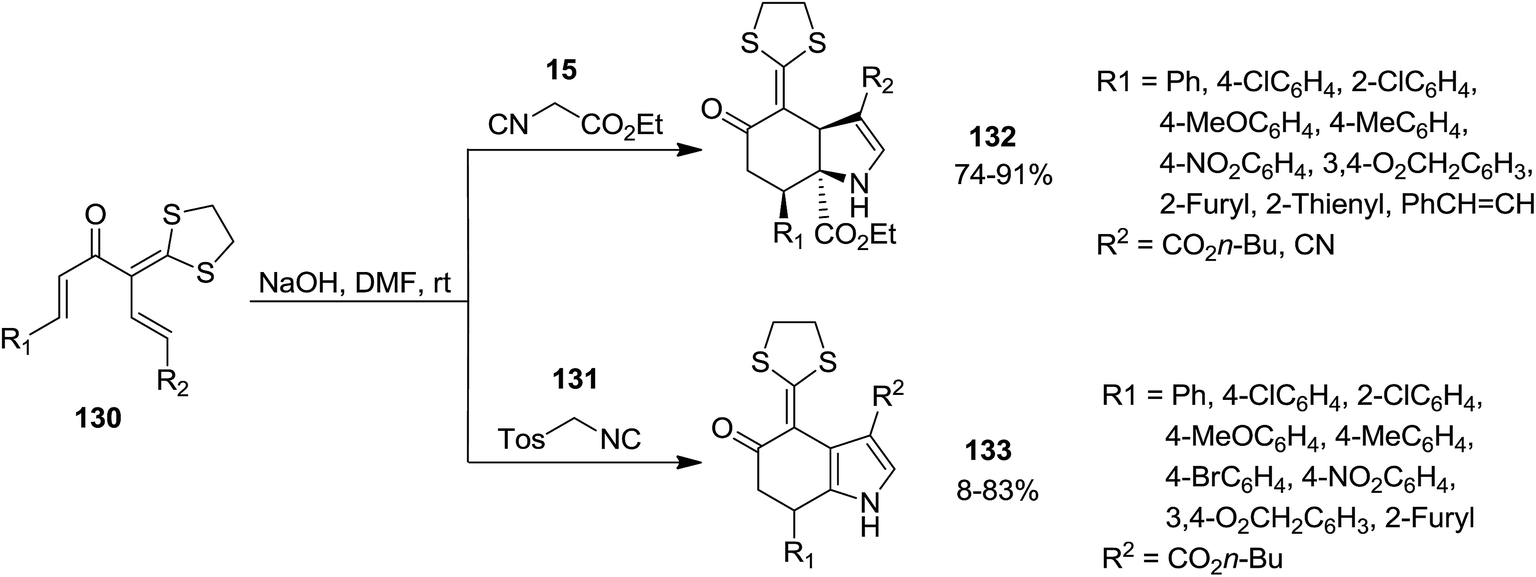
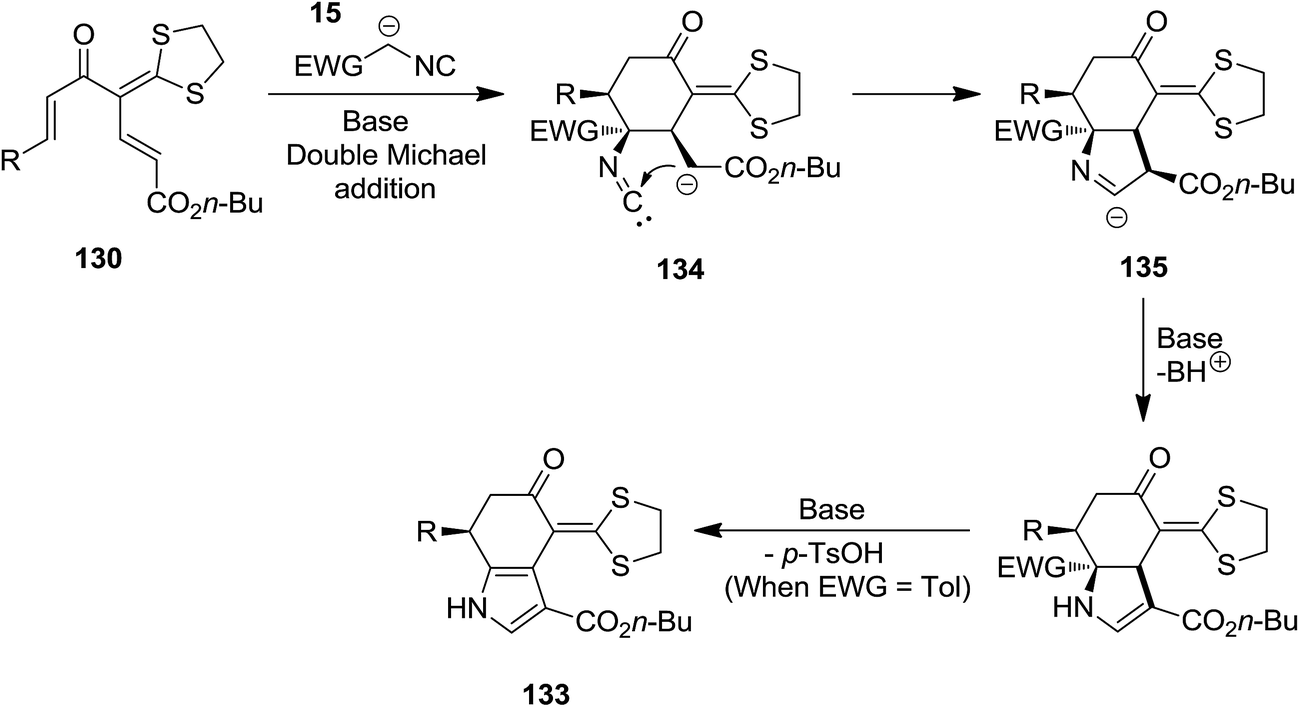
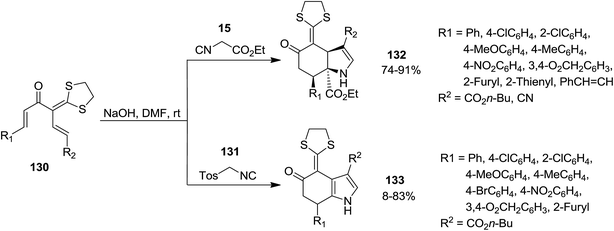
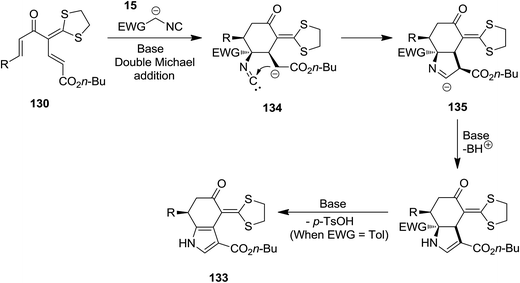
High functionalized tetrahydroindolone 132 as well as dihydroindolone derivatives 133 were obtained in high yields via a base-promoted sequential reaction which involved [5 + 1] cyclization followed by generation of pyrrole ring (Scheme 28). Reaction of 1,5-dielectrophilic substrates 130 with activated methylene isocyanides 15 led to the formation of corresponding tetrahydroindolone 132 while using tosylmethyl isocyanide 131 resulted in dihydroindolones 133.174 The proposed mechanism for the formation of these compounds is depicted in Scheme 29. Evidently, cyclohexanone anion intermediate 134 is generated via diastereoselective double Michael addition of ethyl isocyanoacetate 15 (or TosMIC 131) to 1,5-dielectrophile 130. Next, the intramolecular annulations of anion onto the isocyanide carbon followed by protonation, resulted in isomerization providing tetrahydroindolone 135. Dihydroindolones can be obtained by further elimination of tosylic acid.
 |
| | Scheme 28 | |
 |
| | Scheme 29 | |
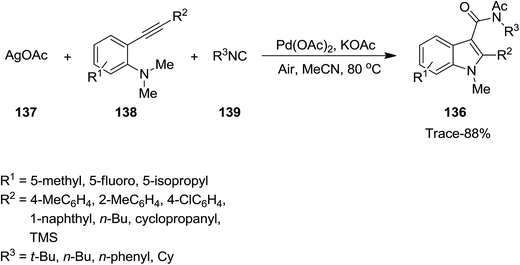
Various 3-amidylindole derivatives 136 were obtained from multi-component reaction of silver acetate 137, 2-alkynylanilines 138 and isocyanides 139 in good yields (Scheme 30). The reaction was catalyzed by palladium using AgOAc which serves as both reactant and oxidant. It is worth noting that replacing AgOAc with other Ag salts did not result in the desired products. It was claimed that intermolecular cyclization and isocyanide insertion results in the formation of all new bonds in a one-pot fashion.175
 |
| | Scheme 30 | |
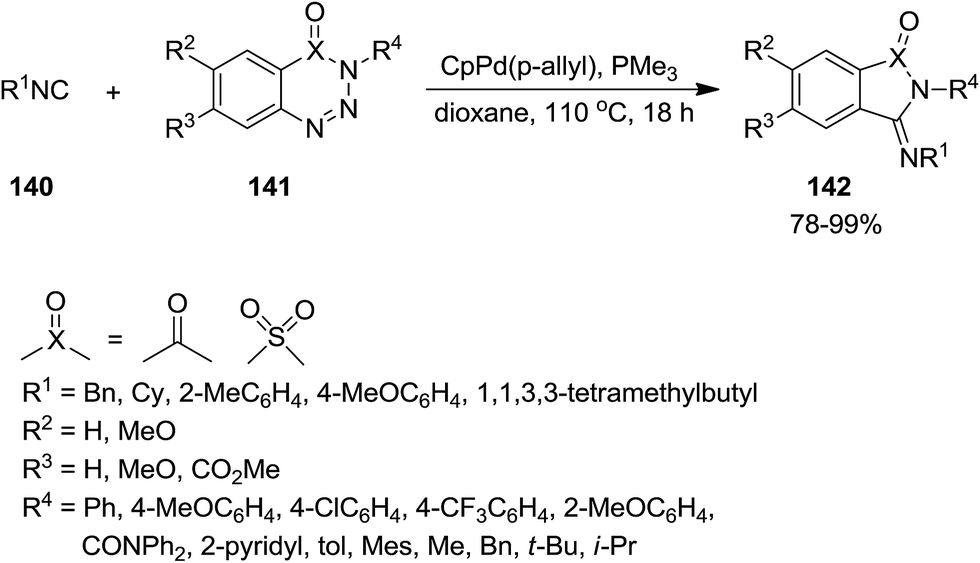
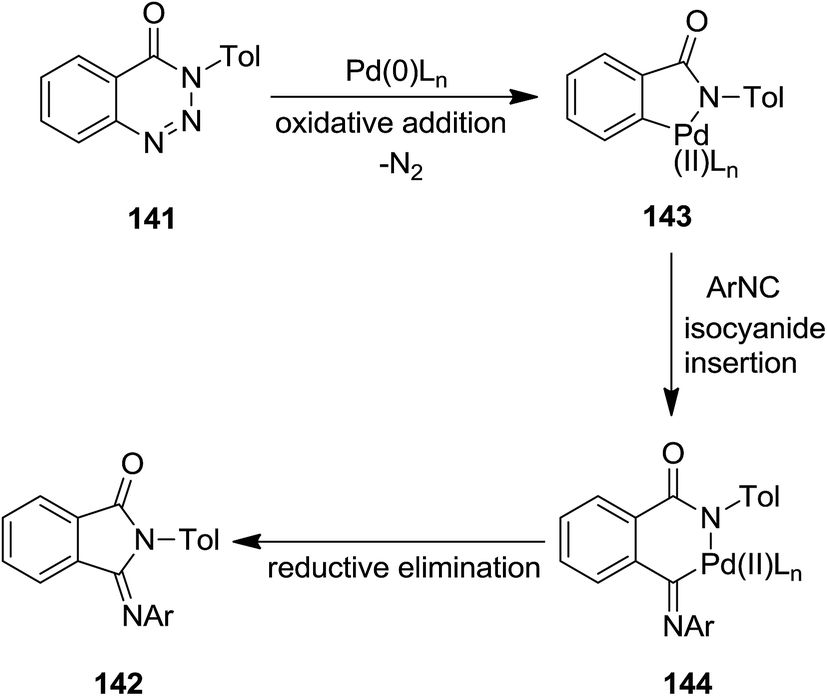
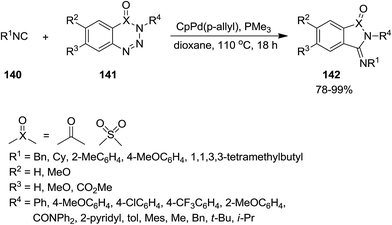
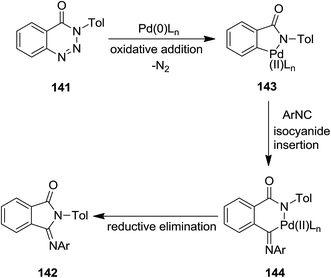
CpPd(π-allyl)/PMe3-catalyzed annulation reaction of isocyanides 140 with 1,2,3-benzotriazin-4(3H)-ones or 1,2,3,4-benzothiatriazine 1,1(2H)-dioxide 141 was developed to afford 3-(imino)isoindolin-1-ones and 3-(imino)thiaisoindoline 1,1-dioxides 142 in satisfactory yields (Scheme 31).176 The proposed mechanism was based on the formation of a five membered azapalladacyclic intermediate 143 via oxidative addition of a C(O)N–N bond to Pd(0) and N2 removal, followed by isocyanide insertion to generate a new six-membered azapalladacyclic intermediate 144. The final product could be obtained in high yield through reductive elimination (Scheme 32). It is worth to mention that the reaction did not proceed in the presence of Ni(0)/PMe3 as catalyst.
 |
| | Scheme 31 | |
 |
| | Scheme 32 | |
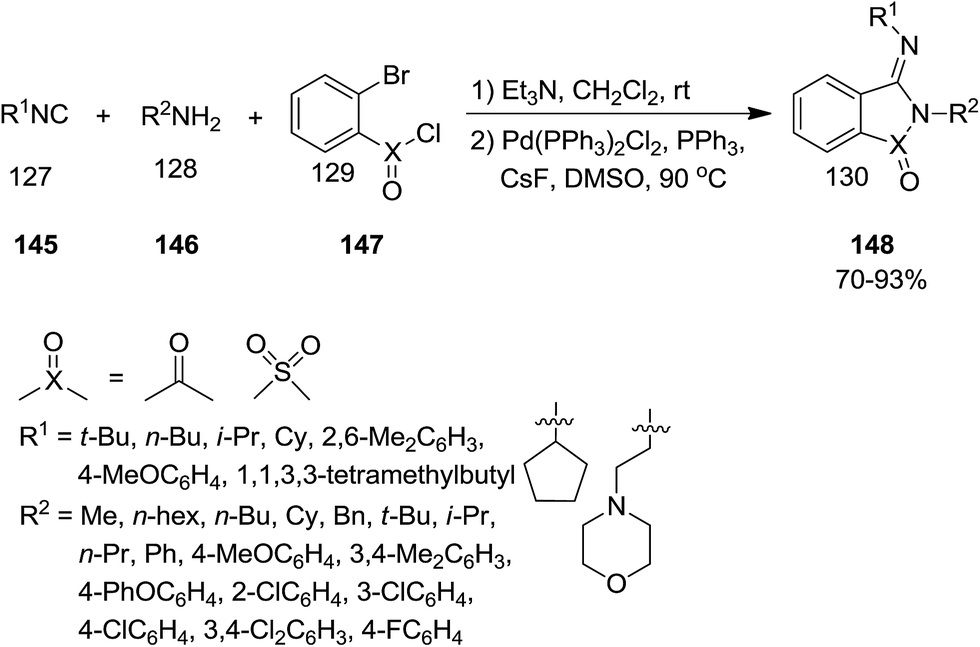
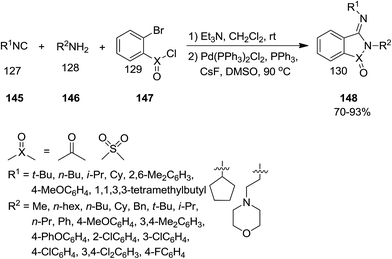
It was reported that Pd could catalyze the annulations of isocyanides 145, amines 146 and 2-bromobenzoyl chloride or 2-bromobenzene-1-sulfonyl 147 to afford (3E)-(imino)isoindolin-1-one and (3E)-(imino)thiaisoindoline 1,1-dioxide derivatives 148 in high yields (Scheme 33).177 Beside high yields, the high stereoselectivity, simplicity, using commercial or easy available starting materials and broad substrate scope were other advantages of this atom economy synthetic approach. This protocol was expanded for the synthesis of phenanthridines and dibenzooxazepines, using 2′-bromo-[1,1′-biphenyl]-2-amine and 2-(2-bromophenoxy)aniline as substrates respectively (Scheme 33).
 |
| | Scheme 33 | |
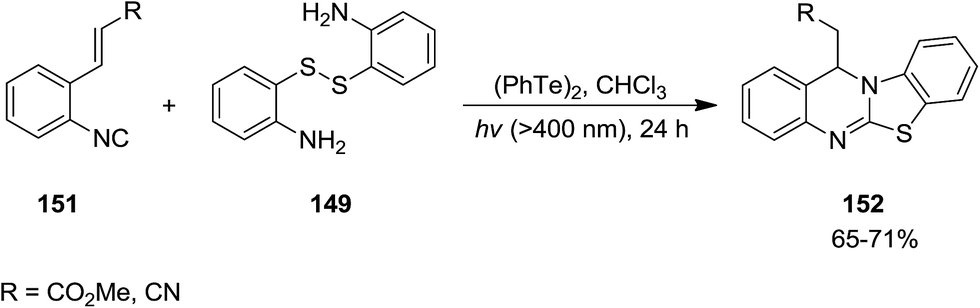
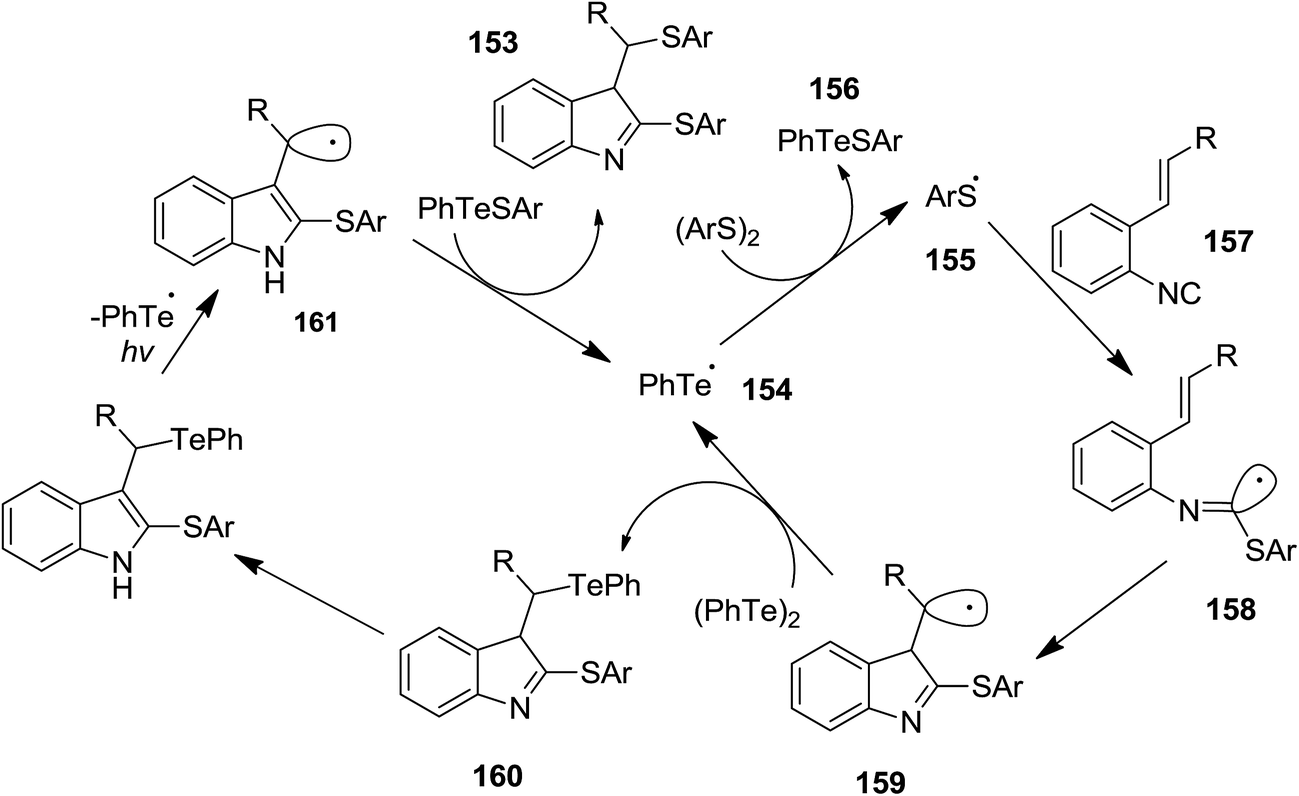
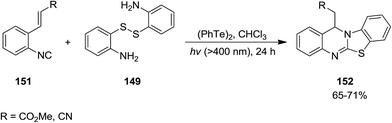
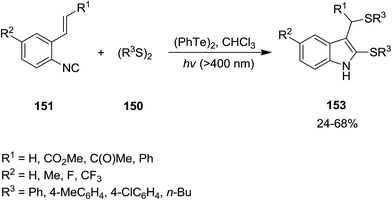
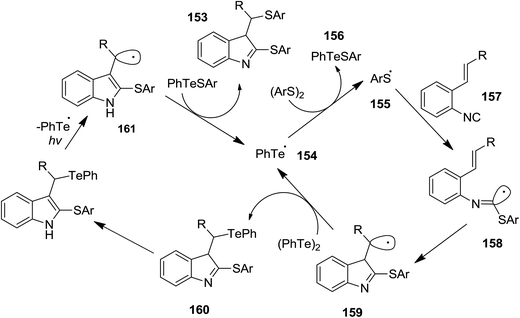
Ogawa et al. reported photoinduced, ditelluride-promoted synthesis of bisthiolated indoles 152 and 153 from reaction of organic disulfides 149 and 150 and o-ethenylaryl isocyanides 151 under visible-light irradiation, with high selectivities (Schemes 34 and 35).81 Using bis(2-aminophenyl)disulfide 149 as organic disulfide reagent led to the formation of tetracyclic compounds 152 which contained both dihydroquinazoline and benzothiazole scaffolds. It was observed that replacement of (PhTe)2 with (PhSe)2 led to a mixture of products, i.e. the acyclic thioselenation product, bisthiolated indole and the selenothiolated indole instead of the desired product. The plausible mechanism is shown in Scheme 36. Initially, PhTe species 154 was formed via photo-induced homolytic dissociation of diphenyl ditelluride. The latter underwent reaction with organic disulfide to afford the ArS species 155 and the thiotelluride 156. Subsequently, an intermediate 157 was generated from reaction of this species with isocyanide which undergoes 5-exo-cyclization to furnish a new radical as an intermediate 158. Formation of indole species 159 proceeded via SH2 reaction with (PhTe)2, followed by tautomerization. The instability of the C–Te bond in this compound led to homolytic cleavage and the generation of a new radical species 160 which furnished the final product by elimination of the ArS group from the thiotelluride.
 |
| | Scheme 34 | |
 |
| | Scheme 35 | |
 |
| | Scheme 36 | |
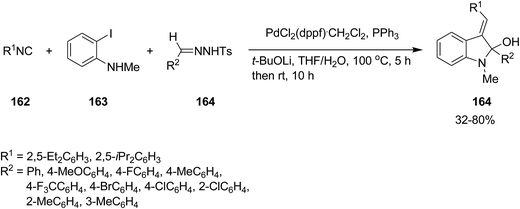
Cheng et al. developed a novel Pd-catalyzed approach based on multi-component reaction of aryl isonitrile 162, N-methyl-2-iodoaniline 163 and N-tosylhydrazone 164 for synthesis of polyfunctionalized indoles 165 (Scheme 37).178 Studying the scope of reaction, it was established that N-methyl and N-ethyl 2-iodoaniline derivatives led to desired product while the reaction did not proceed by using N-phenyl, acetyl, benzyl analogues. This observation was attributed to lower electron density in N atom. Moreover, 2-iodoaniline with electron-donating or electron-withdrawing at phenyl ring could furnish the product. Regarding N-tosylhydrazone, the process exhibited good functional tolerance. Moreover, sterically demanding substrates also provided the products in moderate yields.
 |
| | Scheme 37 | |
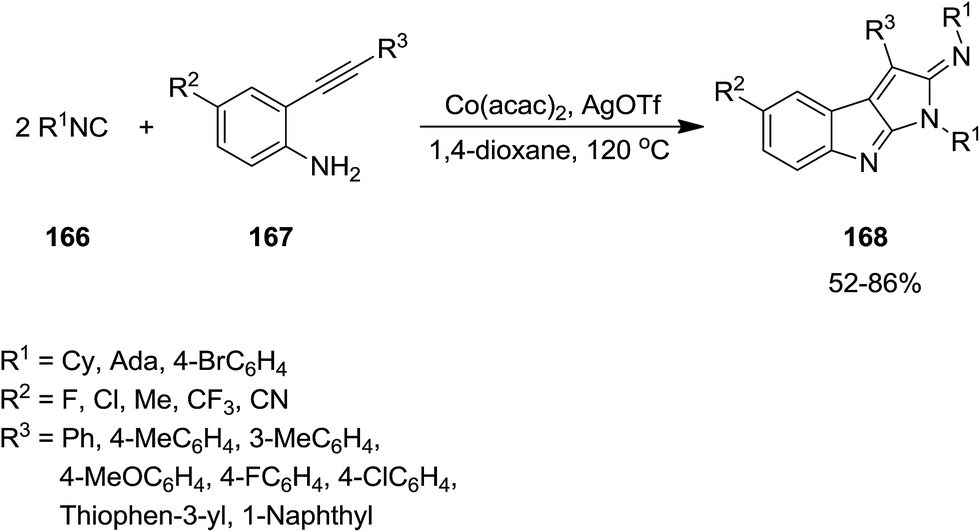
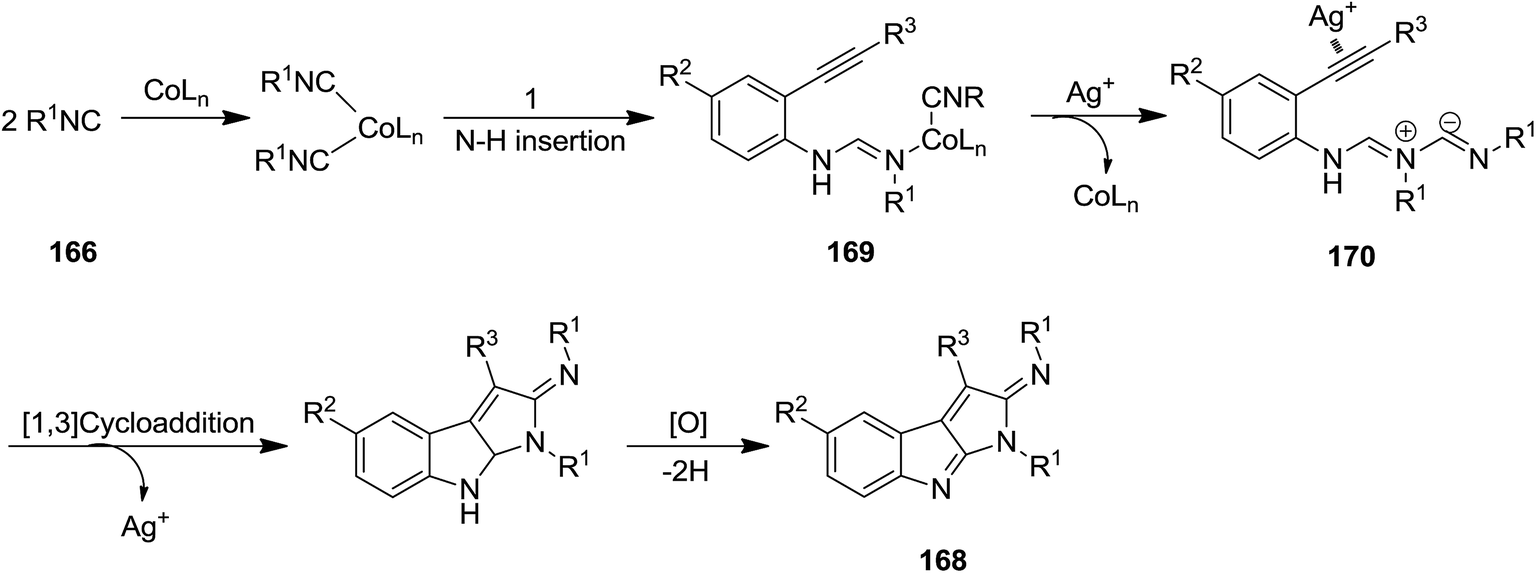
Co(acac)2 and AgOTf were used as catalysts for promoting bicycloaddition of isonitriles 166 and arylethynylanilines 167 to afford substituted pyrrolo[2,3-b]indole 168 (Scheme 38).179 The proposed mechanism was based on formation of cobalt complexes 169 through insertion of the ligated isonitrile into N–H bond of arylethynylanilines 166 followed by second migratory insertion and formation of 170. Subsequently, AgOTf promoted intramolecular 1,3-diplar cycloaddition to furnish 168 (Scheme 39). Good yields, high substrate scope, mild reaction condition and scalability were the advantages of this procedure.
 |
| | Scheme 38 | |
 |
| | Scheme 39 | |
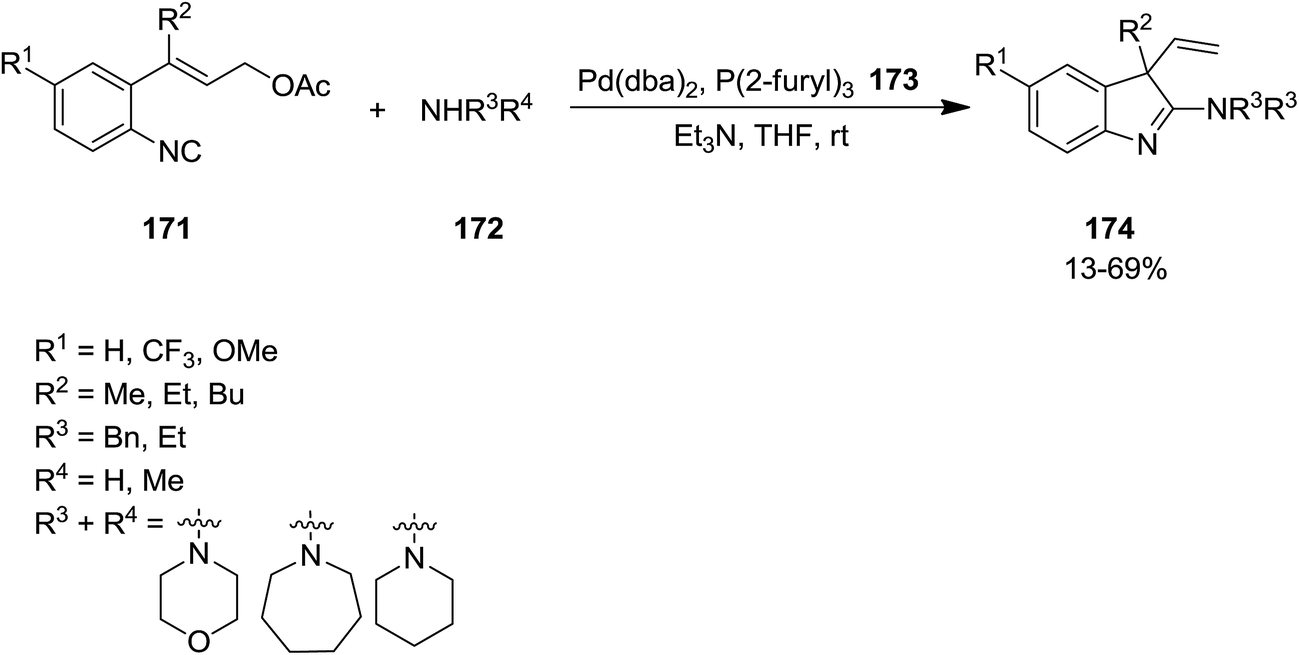
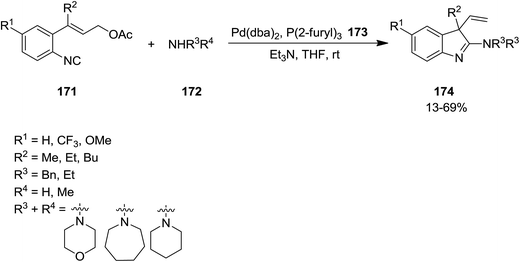
Takemoto et al. developed a novel Pd-catalyzed protocol for synthesis of a series of 3,3-disubstituted 2-aminoindolenine 174 from reaction of isocyanide 171 containing an allyl ester moiety and various amines 172 (Scheme 40).167 The reaction proceeded in the presence of (2-furyl)3 173 as ligand to afford the product in moderate to good yields. Investigating various Pd catalysts revealed that Pd(PPh3)4 was the catalyst of choice.
 |
| | Scheme 40 | |
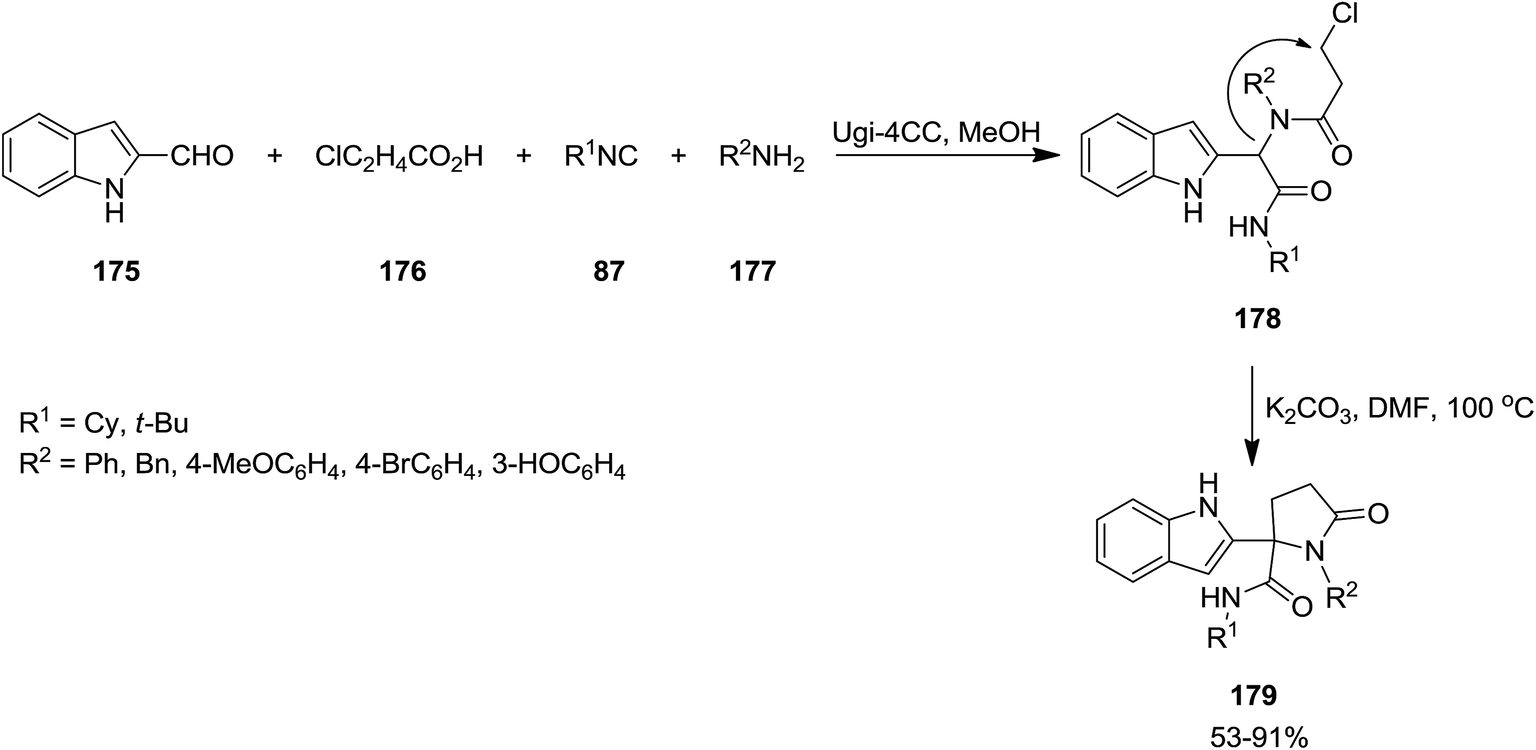
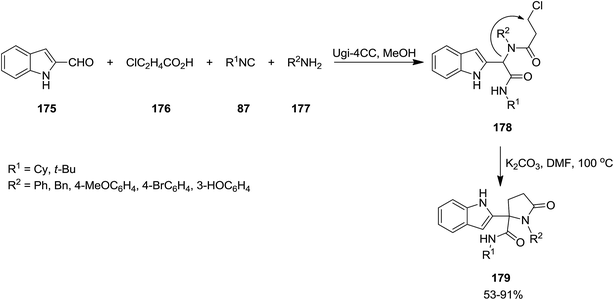
2.2.3 Lactams and lactones. The synthesis of novel derivatives of γ-lactams 179 was developed via cyclization of Ugi-adducts (Scheme 41). In this synthetic strategy, the four-component Ugi-reaction of 2-isocyanides 87, formylindole 175, 3-chloropropanoic acid 176 and amines 177 resulted in the formation of the corresponding Ugi-adduct 178. The latter is subjected to annulation under basic conditions, i.e. K2CO3.180 The scope of this protocol was investigated by using various Ugi-adducts. Moderate to high yields, mild reaction conditions, relatively low reaction times and using cost-effective base were the advantages, mentioned for this strategy.
 |
| | Scheme 41 | |
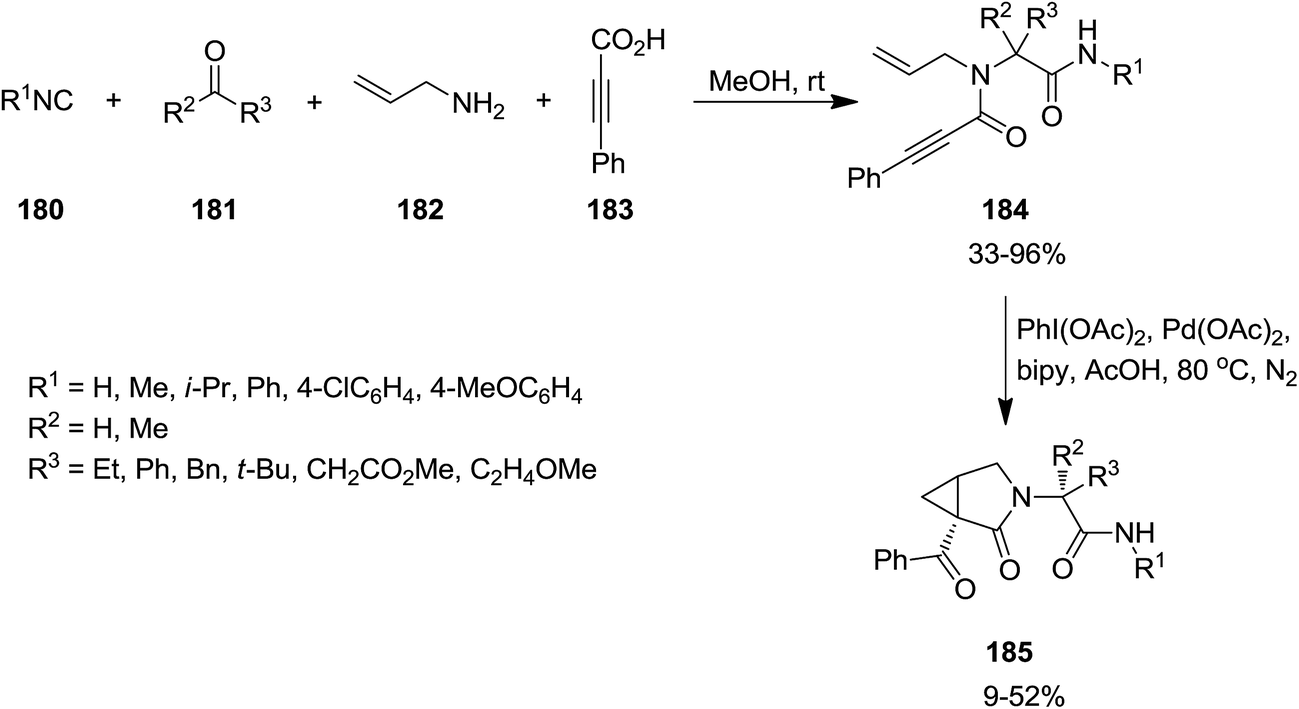
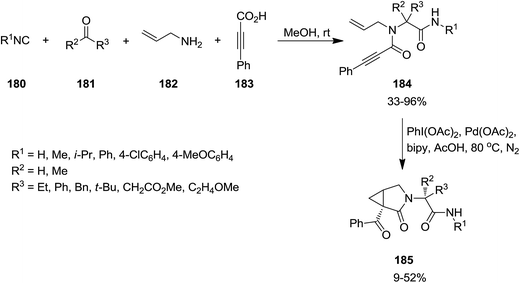
Four-component Ugi reaction of isocyanide 180, aldehydes or ketones 181, amine 182 and acetylene 183 was exploited for the synthesis of a series of 1,6-enynes 184 which subsequently underwent PdII/IV catalytic oxidative cyclization to afford N-functionalized 3-aza-bicyclo[3.1.0]hexan-2-one derivatives 185 in moderate yields (Scheme 42).181 In some cases microwave irradiation was used to reduce the reaction times and subsequent formation of polymer and some side products. The best and worst results were observed for benzyl isocyanide and phenyl isocyanide respectively. It is worth noting that the presence of 2,2′-bipyridyl as an additional ligand is essential for the reaction to proceed, since degradation of the starting material was observed due to its absence.
 |
| | Scheme 42 | |
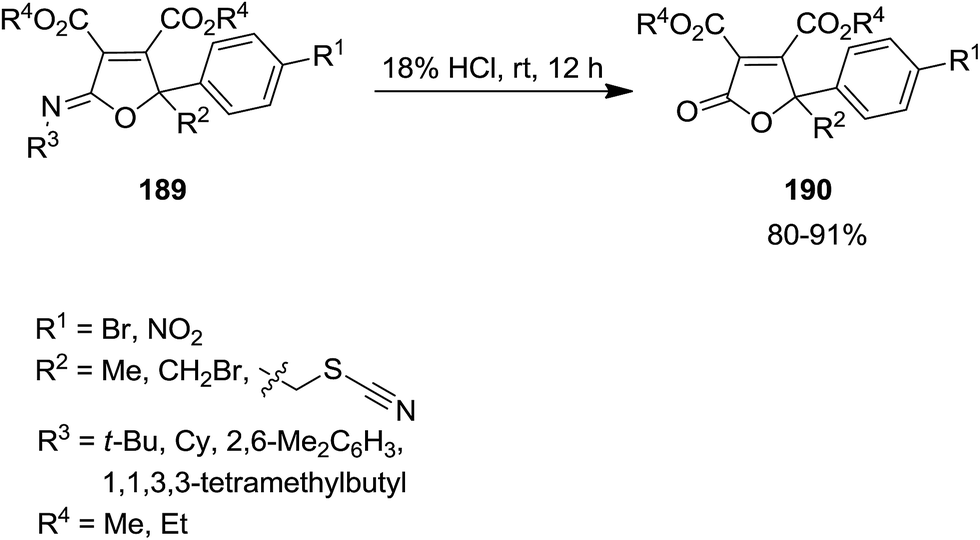
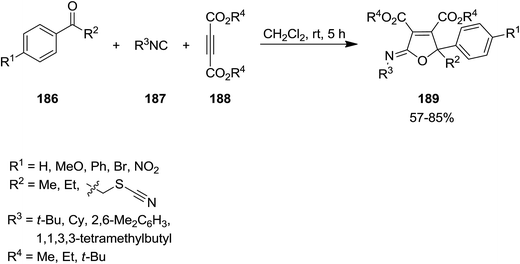
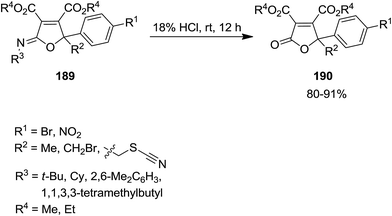
Catalyst-free three component reaction of carbonyl-containing compounds 186, isocyanides 187 and dialkyl acetylenedicarboxylate 188 such as 1-phenyl-2-thiocyanatoethanone derivatives proceeded at room temperature to afford the corresponding poly substituted iminolactones 189 in good yields (Scheme 43).182 The generality of this synthetic strategy was investigated using various isocyanides, dialkyl acetylenedicarboxylates and 1-phenyl-2-thiocyanatoethanone derivatives. It was found that carbonyl compounds with electron-withdrawing groups at α position of CO or para position of Ph group were more reactive. The authors expanded this study to the synthesis of butenolide derivatives 190 via hydrolysis of synthesized iminolactones under acidic conditions (Scheme 44).
 |
| | Scheme 43 | |
 |
| | Scheme 44 | |
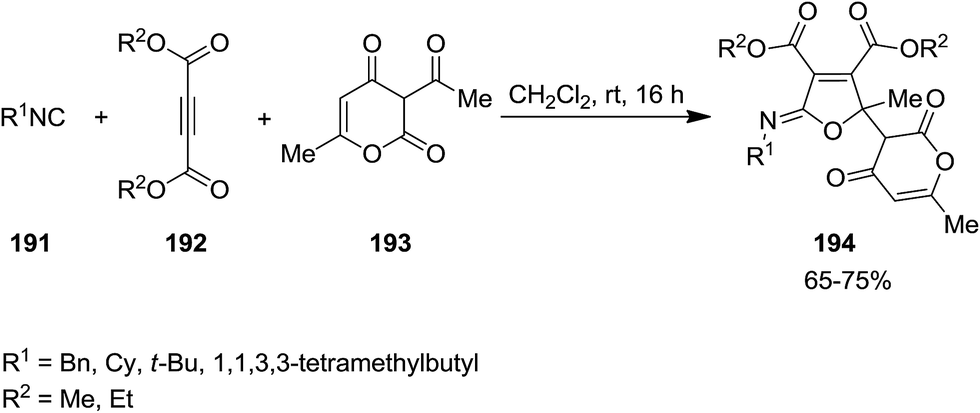
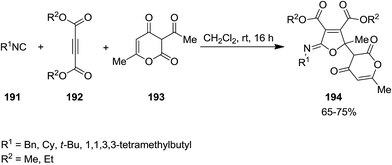
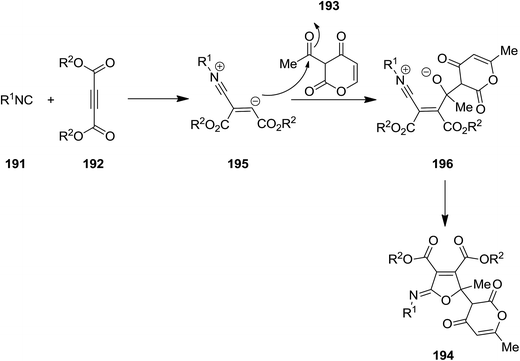
Shaabani et al. developed an isocyanide-based catalyst-free protocol for efficient synthesis of poly-functionalized iminolactone derivatives 194 with pyran–furan moiety from multi-component reaction of isocyanide 191, dialkyl acetylenedicarboxylate 192, and 3-acetyl-6-methyl-3H-pyran-2,4-dione 193 under mild reaction condition (Scheme 45).183 The proposed mechanism was based on formation of zwitterionic intermediate 195 from reaction of dialkyl acetylenedicarboxylate and isocyanide followed by generation of compound 196 through addition of 195 to keto group of 3-acetyl-6-methyl-3H-pyran-2,4-dione. The desired product would be furnished by intermolecular cyclization (Scheme 46).
 |
| | Scheme 45 | |
 |
| | Scheme 46 | |
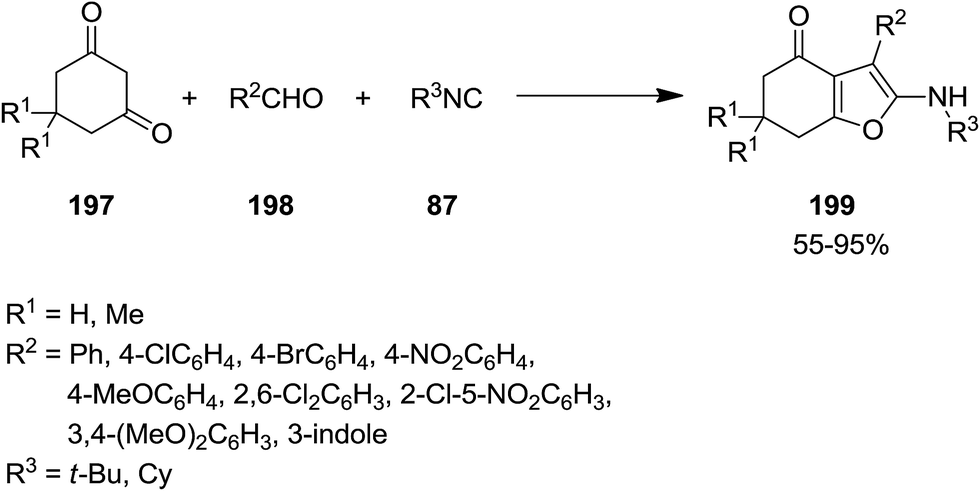
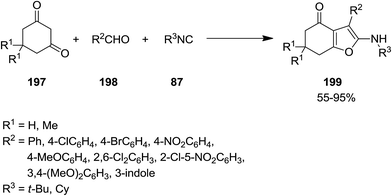
2.2.4 Furans. The application of isocyanides for synthesis of furan derivatives have been reported extensively in the literature.184–186 Exploiting microwave irradiation, un-catalyst three-component reaction of isocyanide 87, cyclic 1,3-diketones 197 and aryl aldehyde 198 was performed to give 6,7-dihydrobenzofuran-4(5H)-one derivatives 199 under solvent-free conditions (Scheme 47).187 It is worth noting that using acyclic 1,3-diketones did not furnish the desired products. Investigating the photophysical characteristics of products established their metal-sensing ability which can be used for developing of chemical sensors. In the presence of Al(III) ions some of the synthesized derivatives showed color-change which could be detected with naked eye.
 |
| | Scheme 47 | |
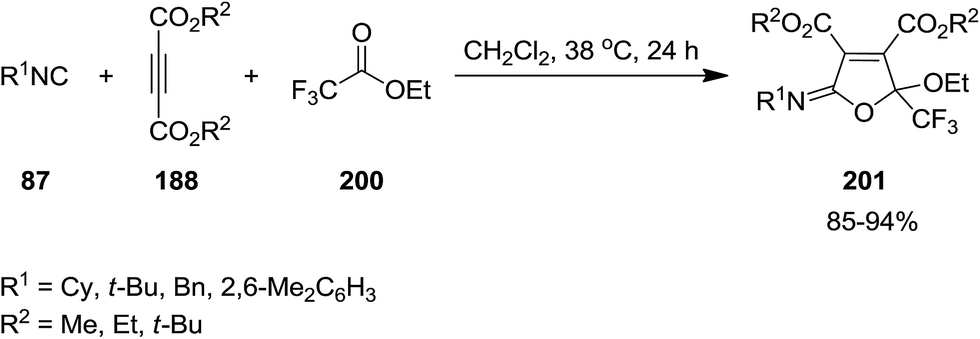
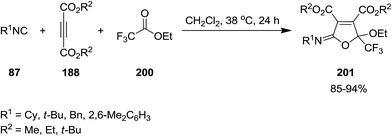
Poly-functionalized furan-based heterocycles, dialkyl 5-[alkyl(aryl)imino]-2-ethoxy-2-(trifluoromethyl)-2,5-dihydrofuran-3,4-dicarboxylates, 201 were synthesized via un-catalyzed multicomponent reaction of dialkyl acetylenedicarboxylates 188, isocyanides 87 and ethyl trifluoroacetate 200 (Scheme 48).188 High yields, mild reaction conditions, broad substrate scope were the advantages of this approach. Furthermore, the reagents could just be mixed for performing the reaction without any modification and activation.
 |
| | Scheme 48 | |
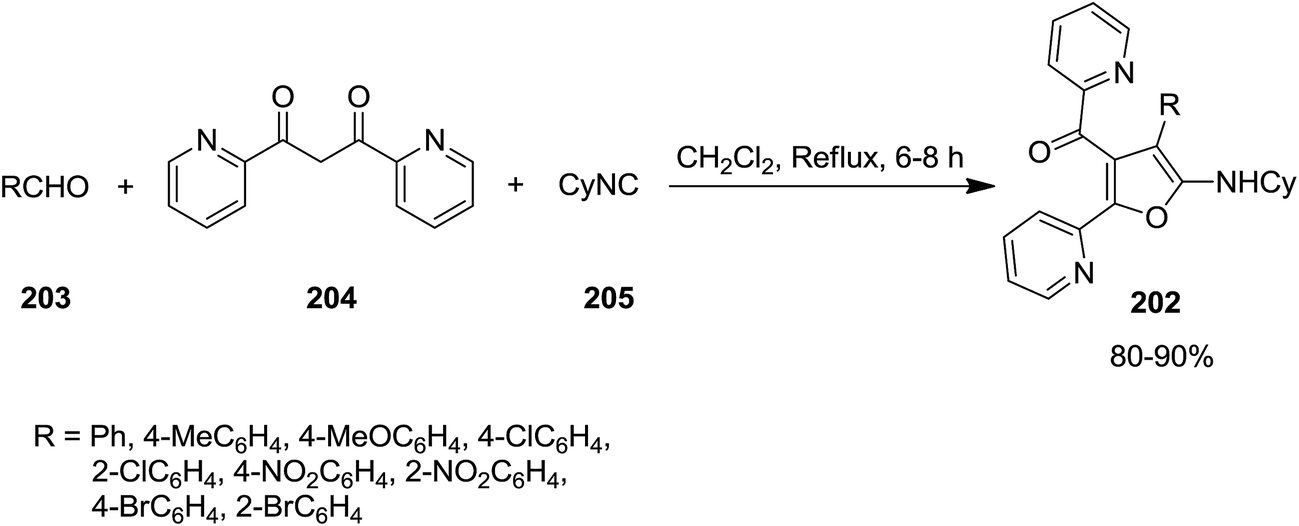
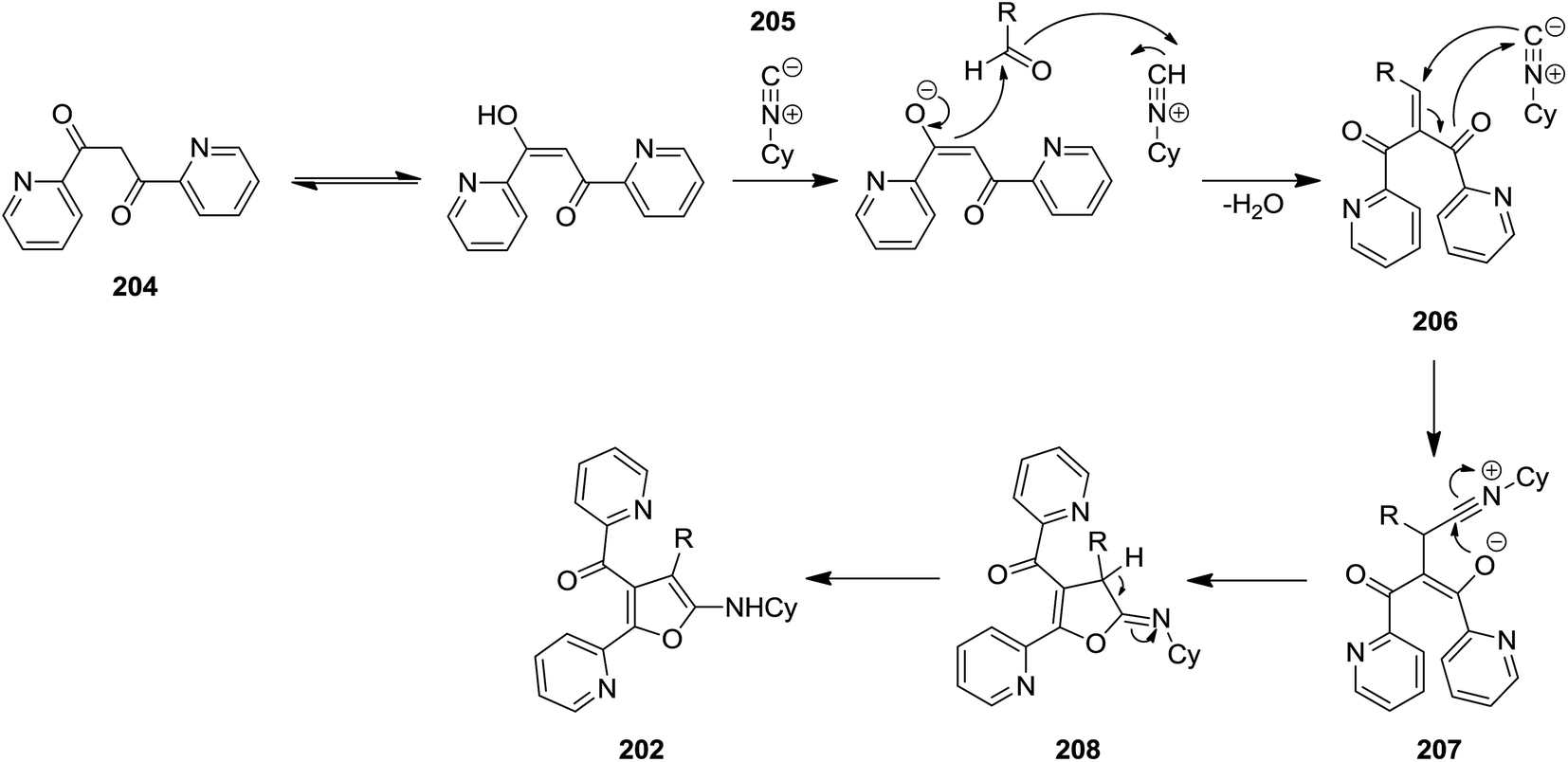
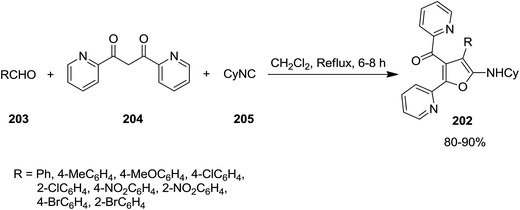
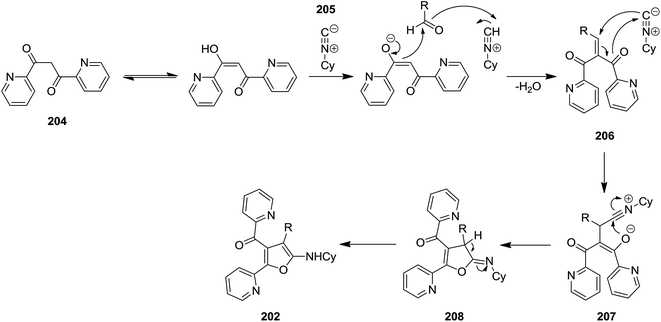
High functionalized furans, [4-(4-aryl)-5-(cyclohexylamino)-2-(pyridin-2-yl)furan-3-yl](pyridin-2-yl)-methanone derivatives 202, were obtained via a one pot, three-component and un-catalyzed reaction of aryl aldehydes 203, 1,3-di(pyridin-2-yl)propane-1,3-dione 204 and cyclohexyl isocyanide 205 (Scheme 49).189 The plausible reaction mechanism was based on formation of an intermediate 206 generated by Knoevenagel condensation of an appropriate 1,3-diketone and an aromatic aldehyde with subsequent isocyanide insertion leading to the generation of a new intermediate 207. The latter upon cyclization formed compound 208. Subsequently the isomerization of obtained intermediate furnished the final product 202 in high yield (Scheme 50).
 |
| | Scheme 49 | |
 |
| | Scheme 50 | |
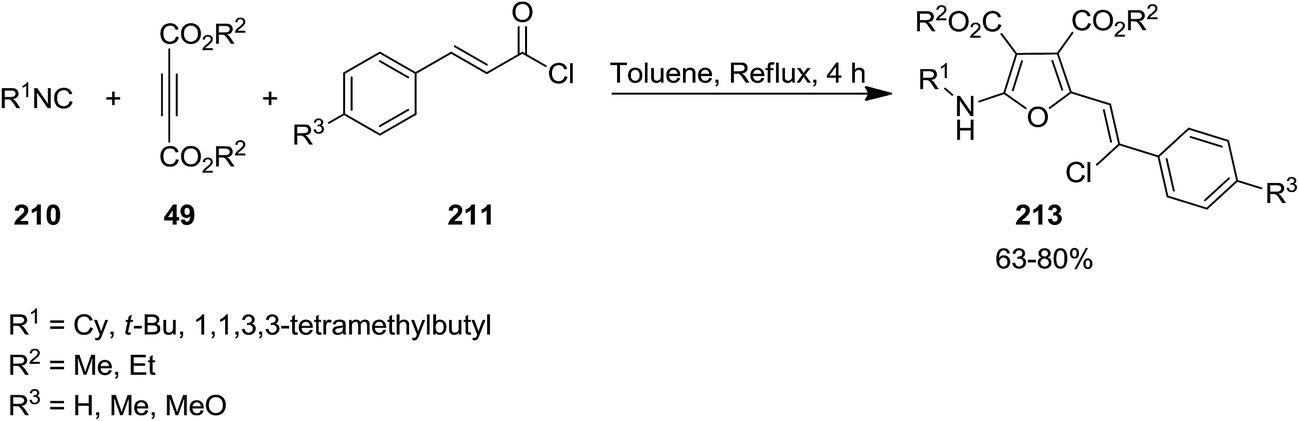
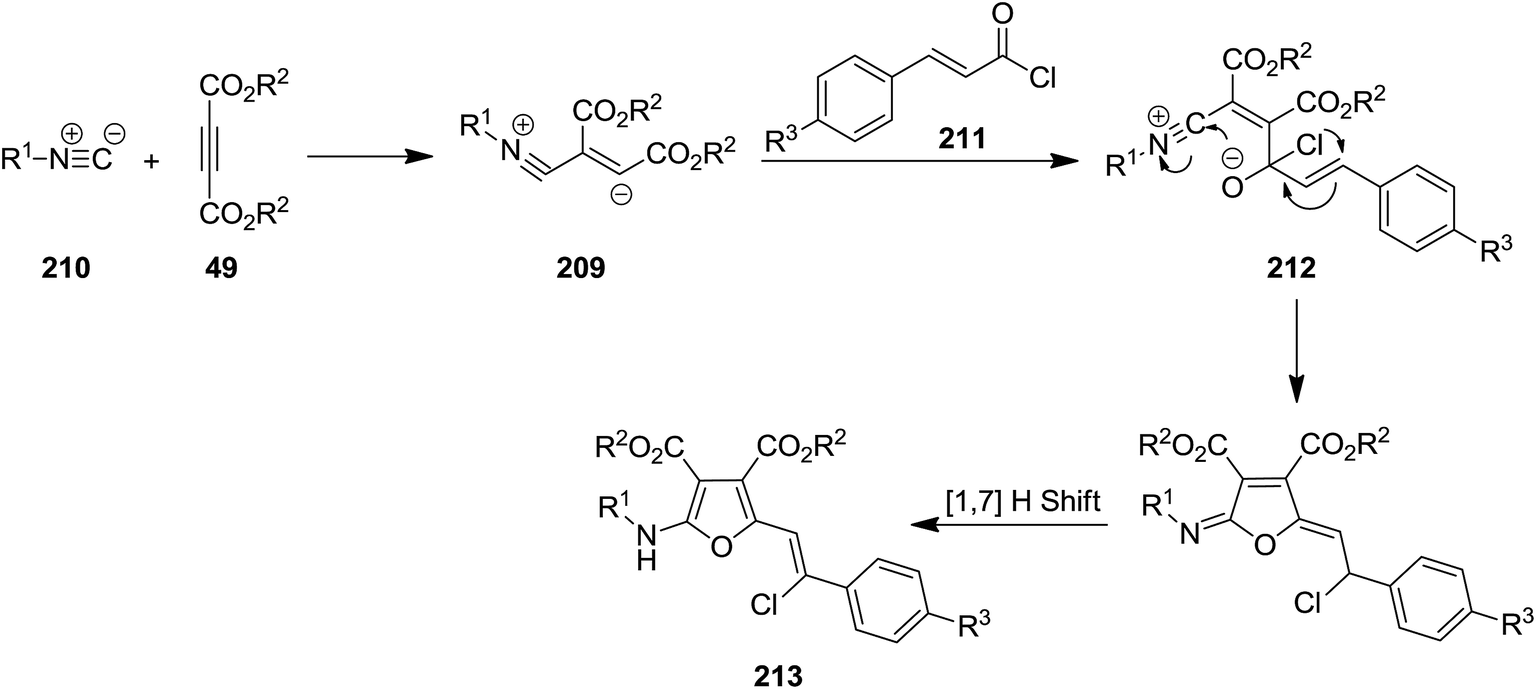
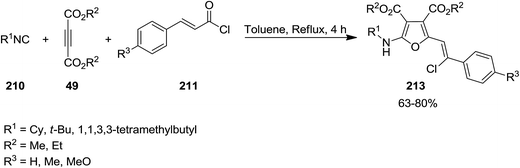
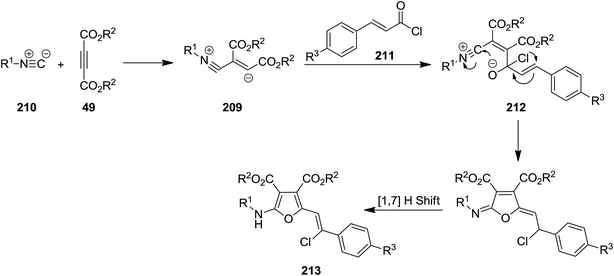
The intermediate 209 derived from reaction of dialkyl acetylenedicarboxylates (DMAD) 49 and isocyanides 210 was trapped with trans-cinnamoyl chlorides 211 to afford an intermediate 212 which subsequently underwent 1,3-migration of Cl along with cyclization to furnish poly-functionalized 2-vinyl furan derivatives 213 in good yields (Schemes 51 and 52).190 To reveal whether Cl migration is intramolecular or intermolecular, a controlled and monitored reaction was performed in the presence of one equivalent of KBr. Obtaining products with no Br substitution proved the intramolecular entity of migration.
 |
| | Scheme 51 | |
 |
| | Scheme 52 | |
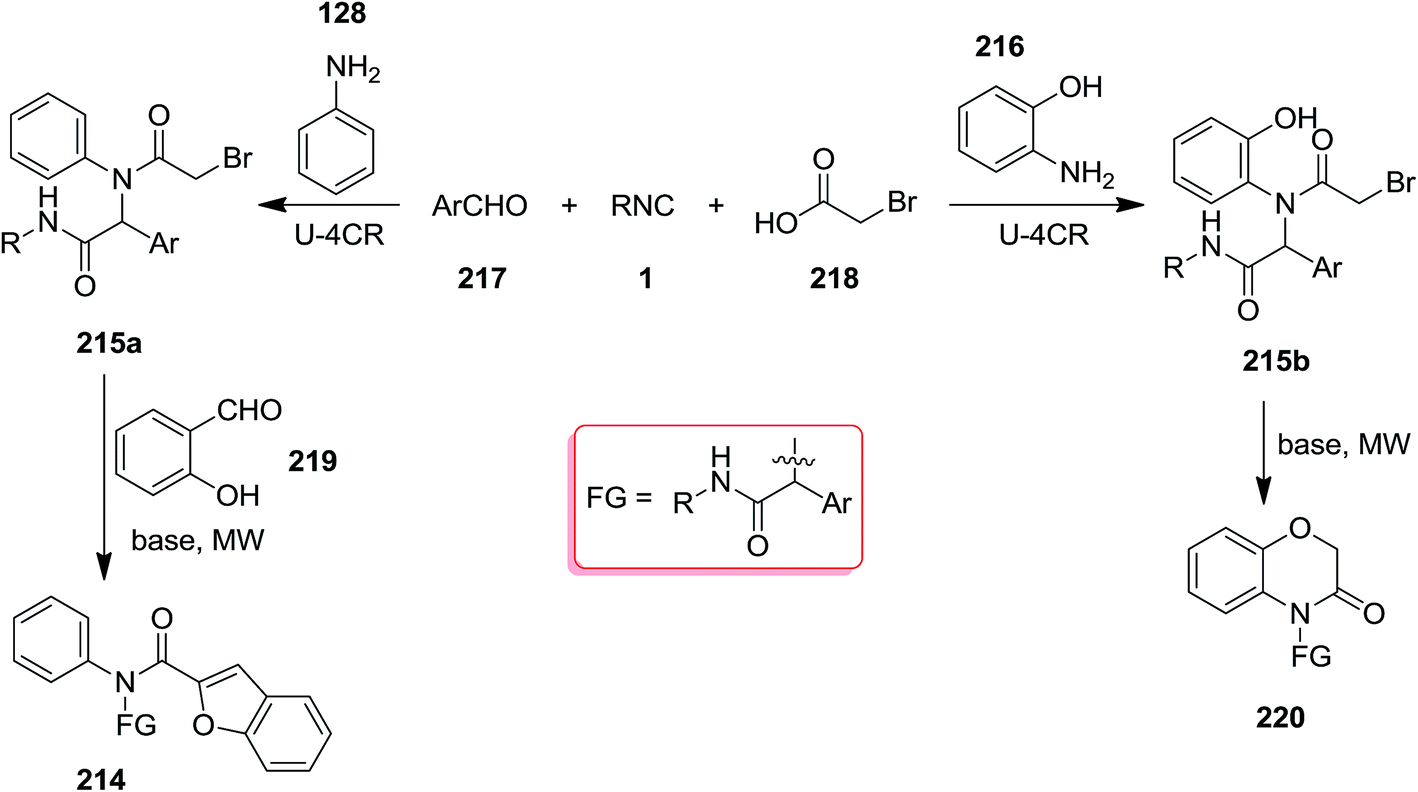
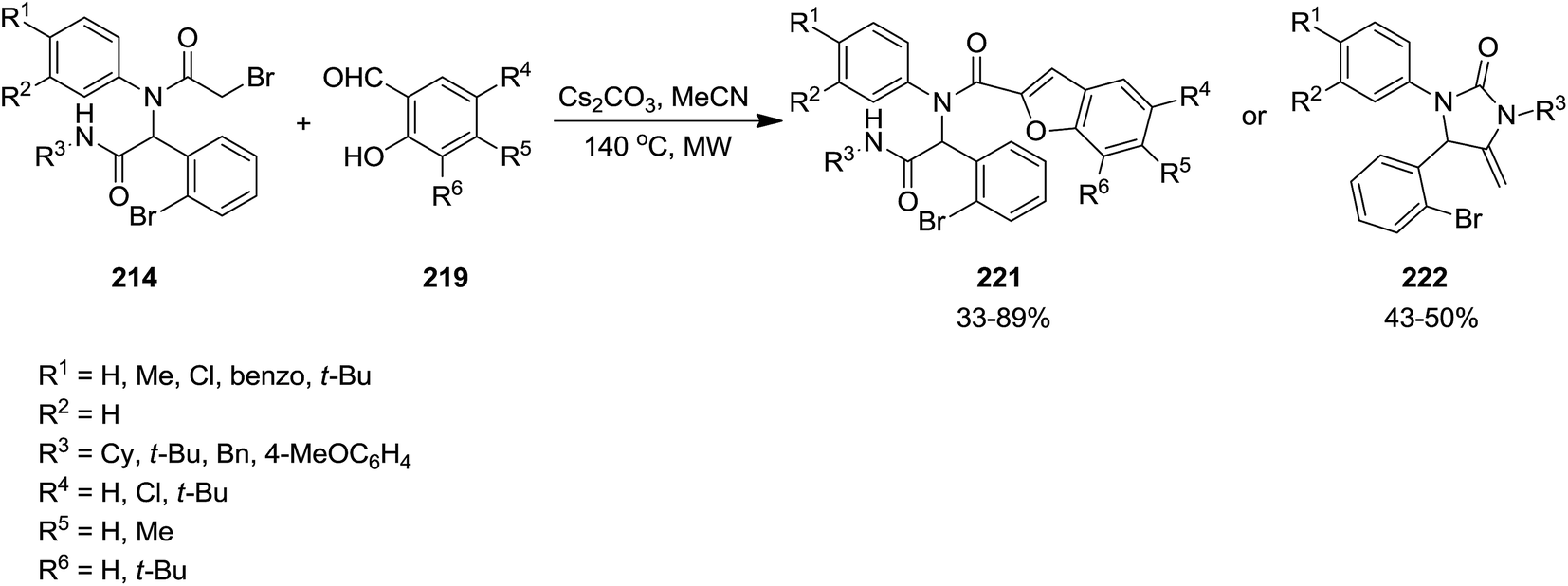
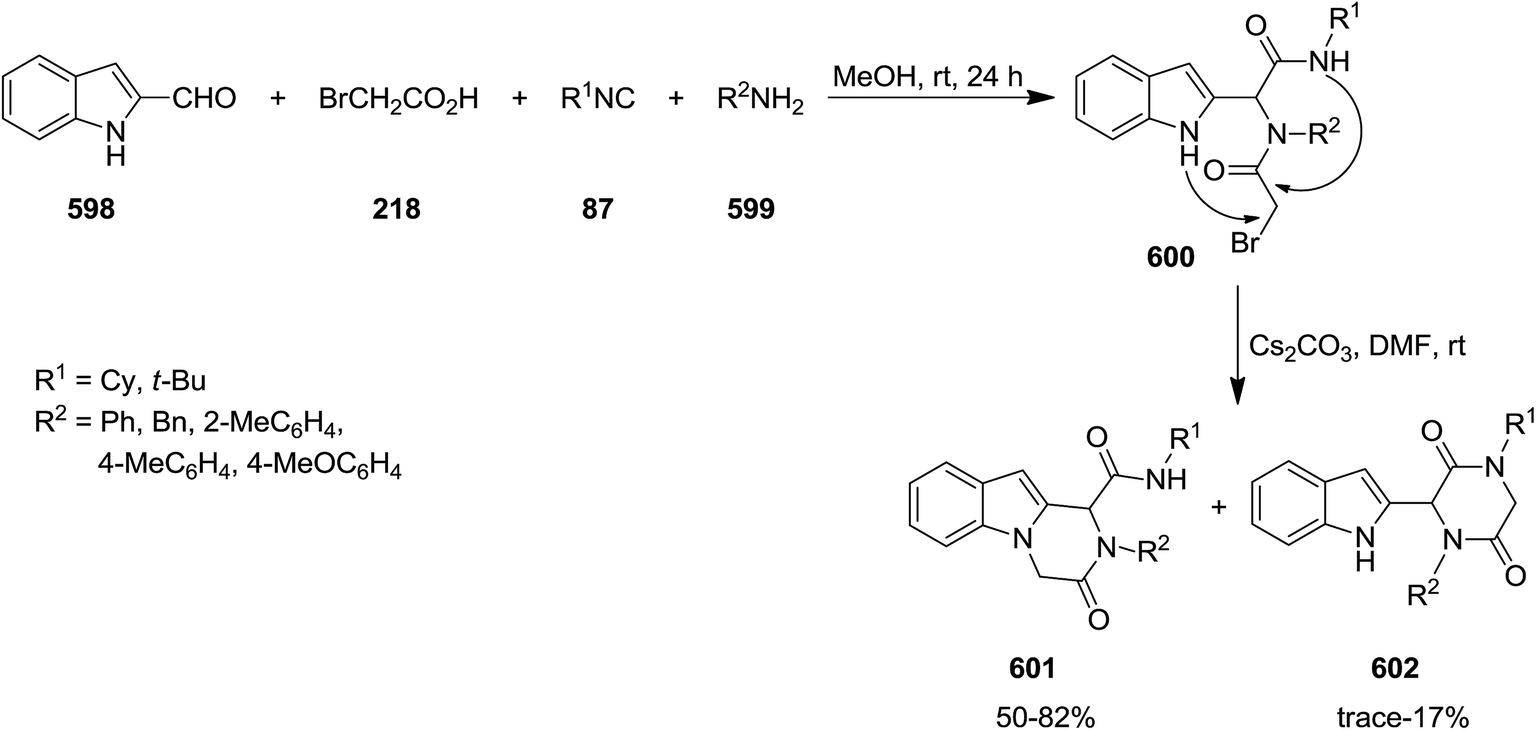
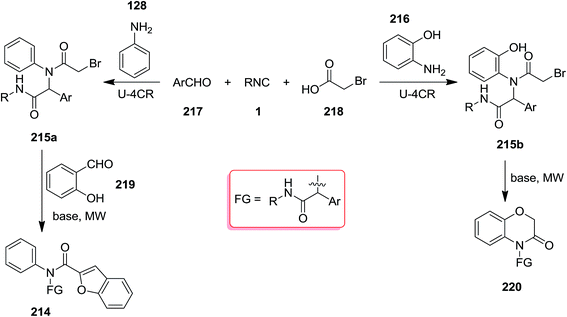
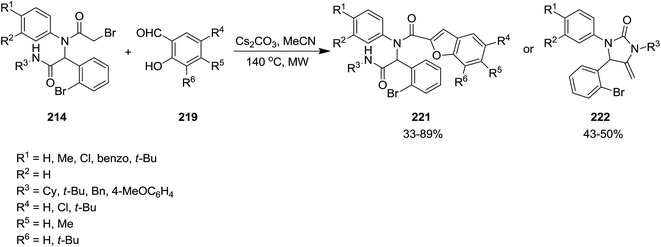
Poly-functionalized benzofuran-2-carboxamide derivatives 214 were obtained in two-step synthetic process. Initially, various derivatives of N-aryl 2-bromoacetamides 215 were obtained from Ugi reaction of aromatic amines 128 or 216, 2-bromobenzaldehyde 217, isocyanides 1 and 2-bromoacetic acid 218 which subsequently transformed into benzofuran-2-carboxamides 214 through Rap–Stoermer reaction with different salicyladehydes 219 under microwave irradiation at 90–140 °C (Schemes 53 and 54).191 It was found that intramolecular N-alkylation of N-aryl 2-bromoacetamides 215 can also be employed for the synthesis of 1,3,4-trisubstituted piperazine-2,5-diones 220. Furthermore, 1,3,5-trisubstituted hydantoins 222, could be obtained via utilizing an electron-deficient salicylaldehyde in the presence of Cs2CO3. 211 could also be obtained from reaction of 214 and 219.
 |
| | Scheme 53 | |
 |
| | Scheme 54 | |
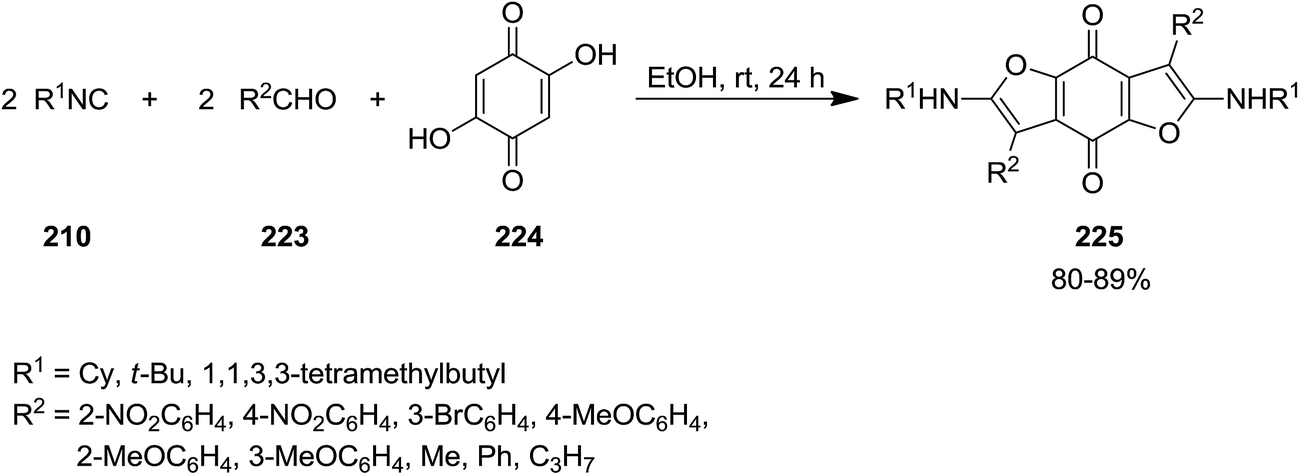
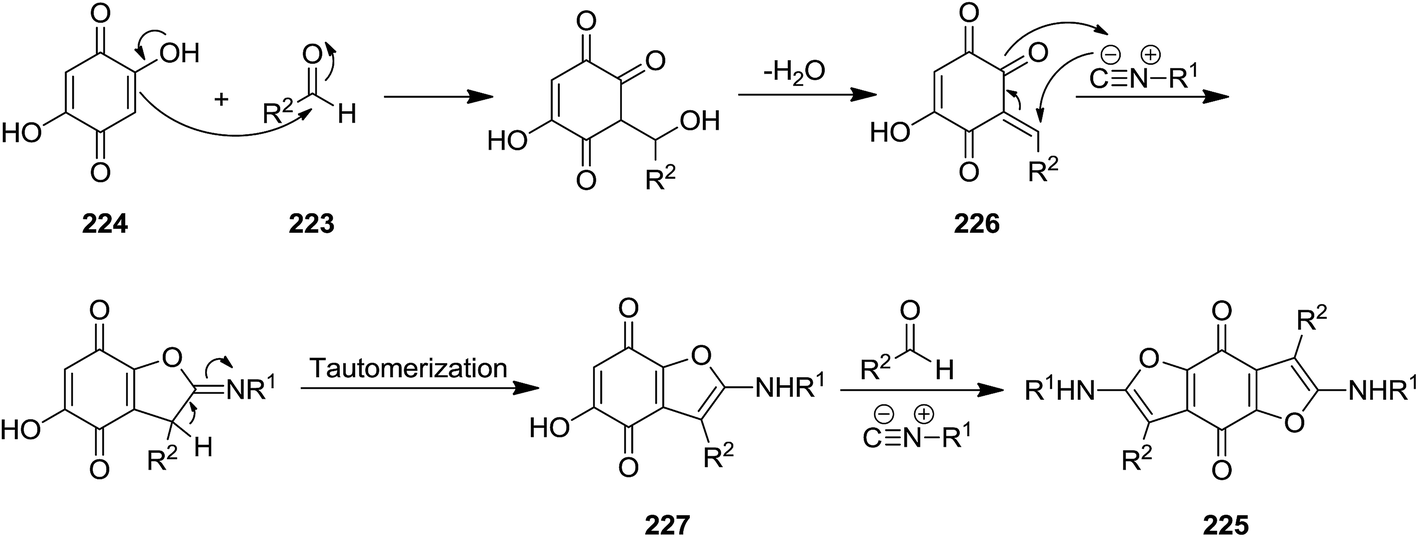
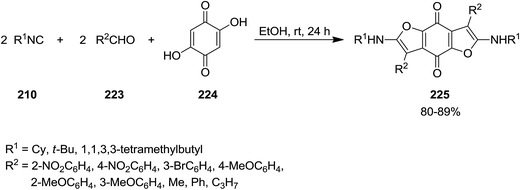
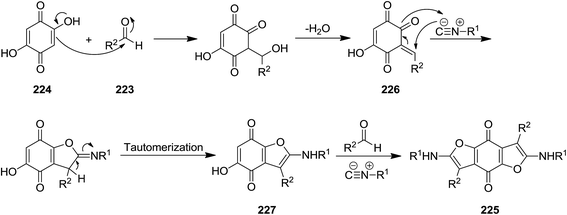
Shaabani et al. reported the utility of isocyanide for efficient synthesis of 2,6-bis(alkylamino)-benzofuro[5,6-b]furan-4,8-diones 225. The synthetic strategy was based on multi-component reaction of isocyanides 210, aldehydes 223, and 2,5-dihydroxycyclohexa-2,5-diene-1,4-dione 224 under mild reaction condition (Scheme 55). The proposed mechanism included initial formation of 226 through reaction of aldehyde and 2,5-dihydroxycyclohexa-2,5-diene-1,4-dione followed by generation of a new intermediate via [1 + 4] cycloaddition reaction with the isocyanides. Compound 227 would be obtained by imine to enamine tautomerization. Repeating the reaction sequence furnished the desired product (Scheme 56). Besides high yields, the process showed broad substrate tolerance and various aldehydes and isocyanides could be used.192
 |
| | Scheme 55 | |
 |
| | Scheme 56 | |
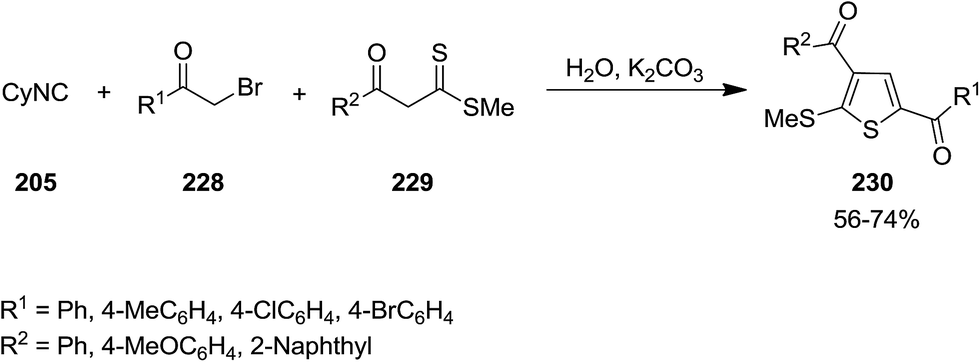
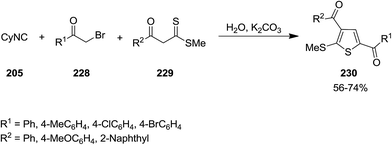
2.2.5 Thiophene. Matloubi Moghaddam et al. developed an environmentally friendly catalyst-free procedure for synthesis of polysubstituted thiophene 230 through multi-component reaction of α-haloketones 228, cyclohexylisocyanide 205 and β-ketodithioesters 229 in water (Scheme 57).193 Broad substrate scope, good yields and facile work-up were the merits of this novel protocol.
 |
| | Scheme 57 | |
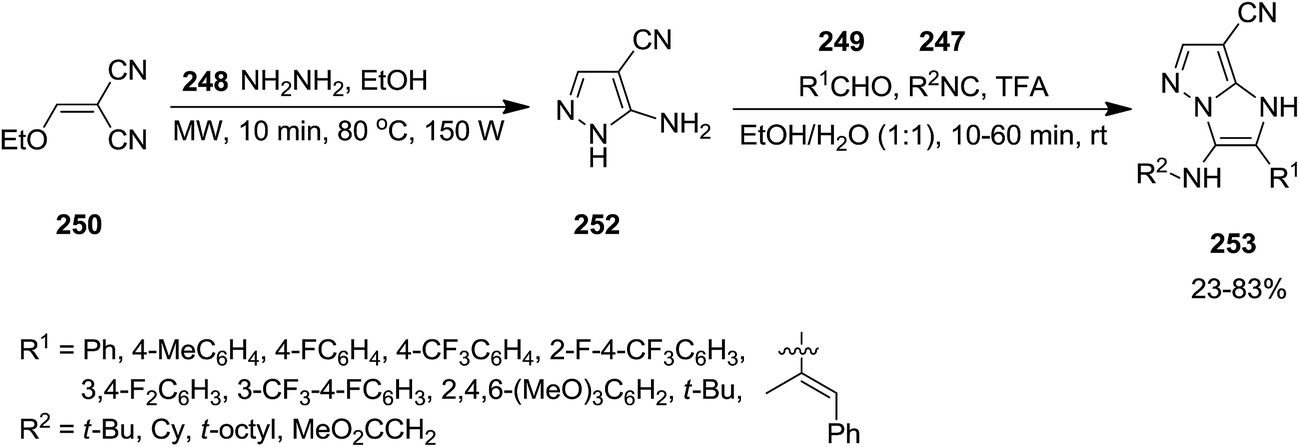
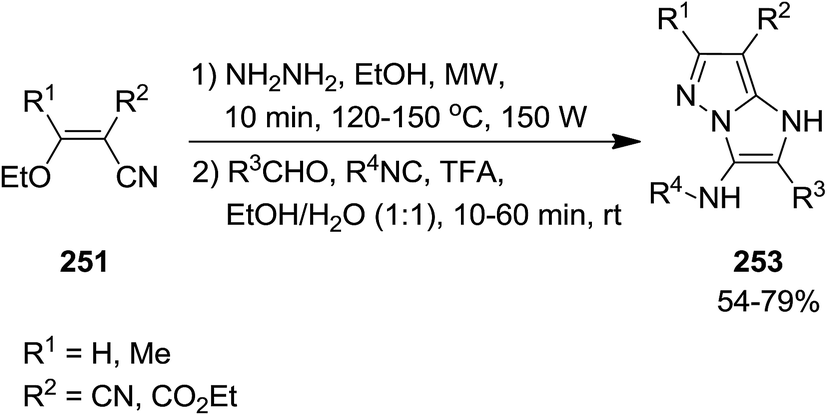
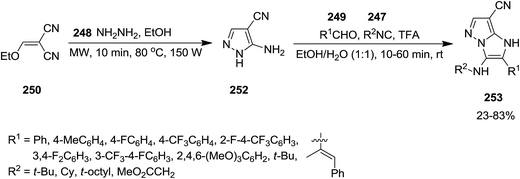
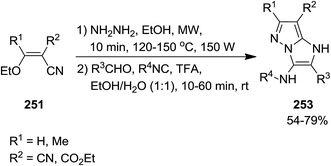
2.2.7 Pyrazole. Using Groebke–Blackburn–Bienaymé reaction, GBB, Kanizsai et al. developed a microwave-assisted, one pot, multicomponent reaction for the synthesis of 1H-imidazo[1,2-b]pyrazole derivatives 253 via reaction of various isocyanides 247, hydrazine hydrate 248, differently substituted aldehydes 249 and ethoxymethylene substituted malononitrile or ethyl cyanoacetate derivatives 250 and 251 (Schemes 63 and 64).198 In this process, 5-aminopyrazole-4-carbonitrile 252 derived via microwave-assisted reaction of hydrazine and ethoxymethylene substituted malononitrile or ethyl cyanoacetate which subsequently subjected to GBB reaction to furnish the desired product. The first step of the reaction should be performed in the absence of water to prevent the generation of a complicated reaction mixture. High yields, short reaction time and broad substrate scope were the advantages, mentioned of this protocol.
 |
| | Scheme 63 | |
 |
| | Scheme 64 | |
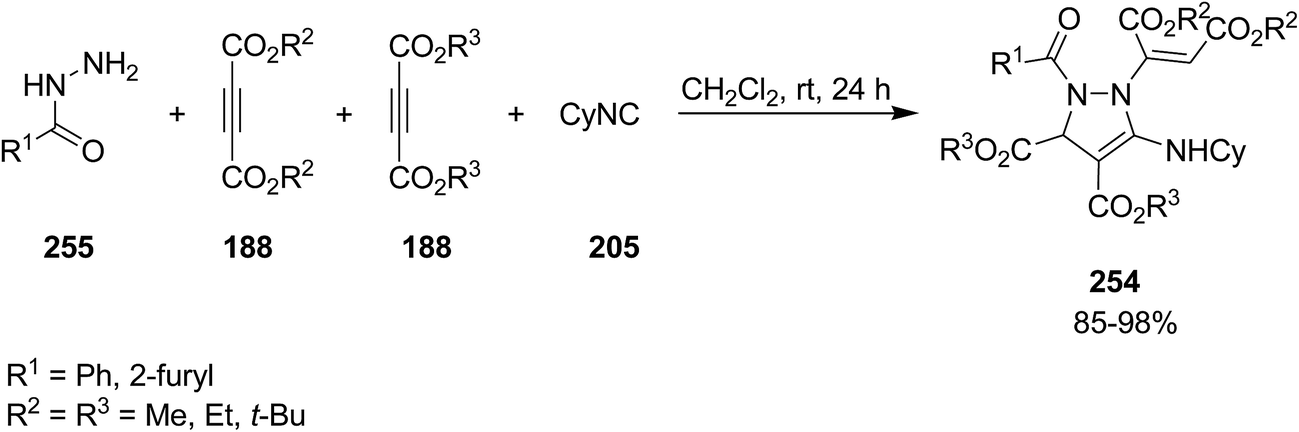
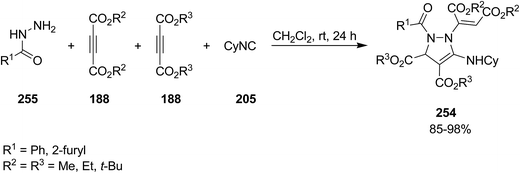
Poly-functionalized pyrazole, dialkyl 1-(1,2-bis-alkoxycarbonylvinyl)-5-cyclohexylamino-2-aroyl-2,3-dihydro-1H-pyrazole-3,4-dicarboxylate 254, were obtained in high yields via a tandem multicomponent reaction of various different arylcarbohydrazides 255, commercially or readily available dialkyl acetylenedicarboxylates 188 and isocyanide 205, (Scheme 65).199 The plausible mechanism involves the addition of cyclohexyl isocyanide to acetylene diester to afford a zwitterions intermediate 256 which can be protonated by an adduct 257 generated from Michael addition of arylcarbohydrazide to acetylene diester to furnish nitrilium cation 258. Next Michael addition of the conjugate base 259 of so-called adduct to nitrilium cation resulted in ketenimine 260, which subsequently cyclized to afford the desired product (Scheme 66).
 |
| | Scheme 65 | |
 |
| | Scheme 66 | |
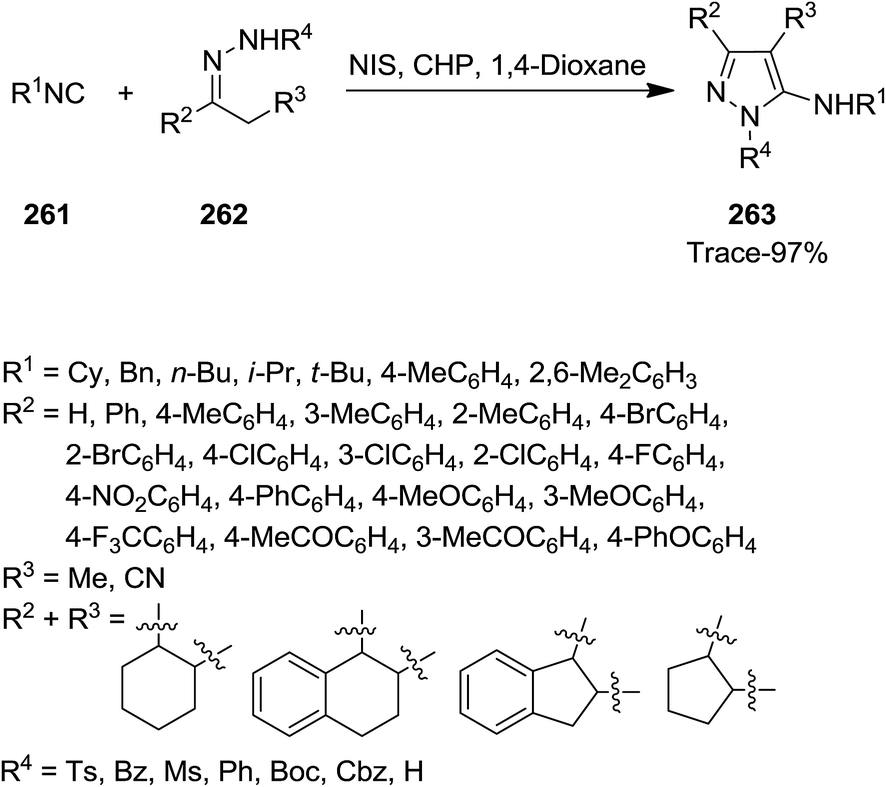
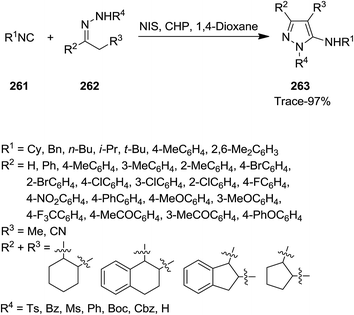
Ji et al. developed a novel, atom efficient protocol for synthesis of aminopyrazole derivatives 263 based on C–C/C–N bond formation via [4 + 1]-cycloaddition of isocyanides 261 with 1,2-diaza-1,3-dienes 262 (Scheme 67). The reaction proceeded via metal-free in situ formation of 1,2-diaza-1,3-dienes. The reaction was mediated by catalytic amounts of N-iodosuccinimide (NIS)/cumene hydroperoxide (CHP).200 The generality of reaction was studied by using substrates with various electron-donating and electron-withdrawing functional groups. The results indicated that reaction of N-tosylhydrazone derivatives and tert-butyl isonitrile led to desired products in moderate to excellent yields. In the case of rigid functionalized N-tosylhydrazones low yields were obtained. N-Substituted hydrazones, however, furnished the desired products in moderate yields. The authors also examined the scope of isocyanides. Benzyl isocyanide led to moderate yields while other isocyanides resulted in products in lower yields.
 |
| | Scheme 67 | |
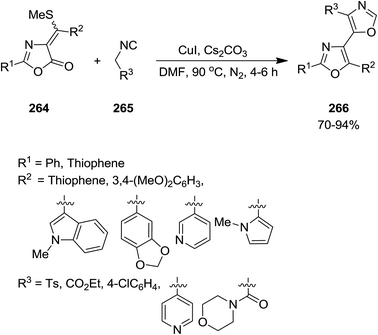
2.2.8 Oxazoles. A novel domino procedure was developed for the synthesis of multi functionalized 4,5′-bisoxazole derivatives 266 from the reaction of 2-phenyl- and 2-(2-thienyl)-4-[(aryl/heteroaryl)-methylene]-5-oxazolones 264 and activated methylene isocyanides 265 in the presence of CuI/Cs2CO3 as catalytic system (Scheme 68).201 The mild reaction conditions, good to excellent yields and the possibility of using wide range of substrates were the advantages of this process. The authors believed that this synthetic method could be used in various research areas such as combinatorial and solid phase synthesis. Furthermore, it could trigger further applications of new bisoxazole derivatives.
 |
| | Scheme 68 | |
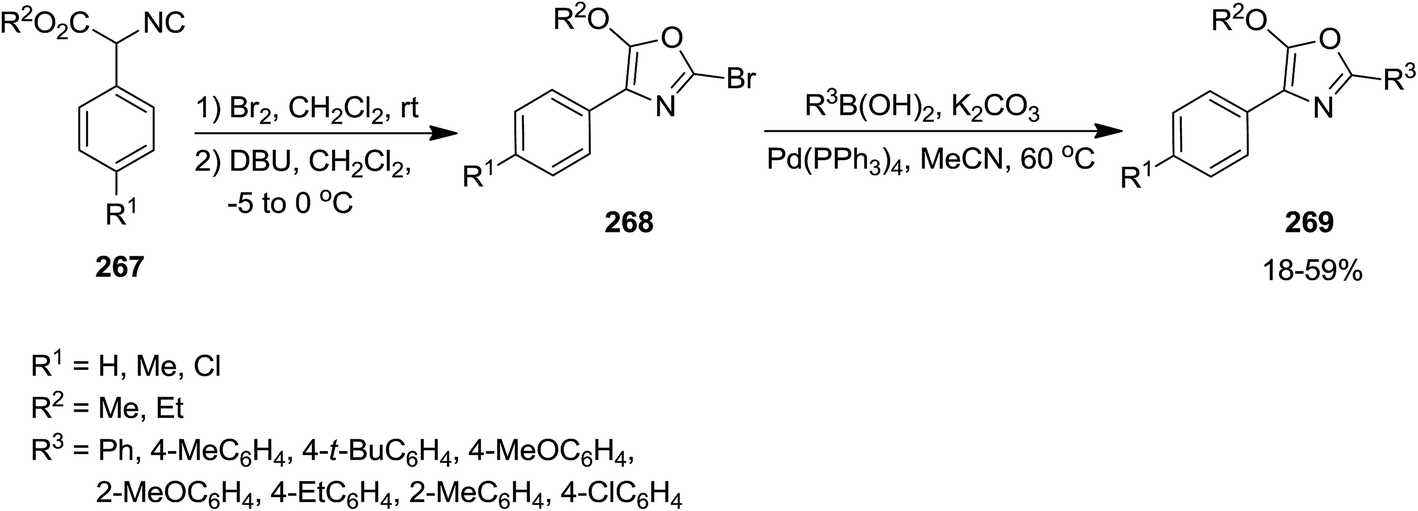
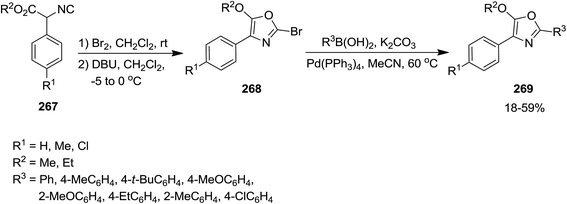
Using isocyanide chemistry, poly-substituted oxazole derivatives 269 were obtained via a two steps sequential process, including bromination/cyclization and a Suzuki cross-coupling reaction (Scheme 69).202 In the first step, ethyl α-phenyl-α-isocyanoacetate 267 reacted with Br2 in dichloromethane at room temperature to afford an intermediate which transformed into bromooxazole 268 at 0 °C in the presence of DBU. This product was unstable and could be submitted to Pd-catalyzed Suzuki–Miyaura cross-coupling reaction to furnish the final product.
 |
| | Scheme 69 | |
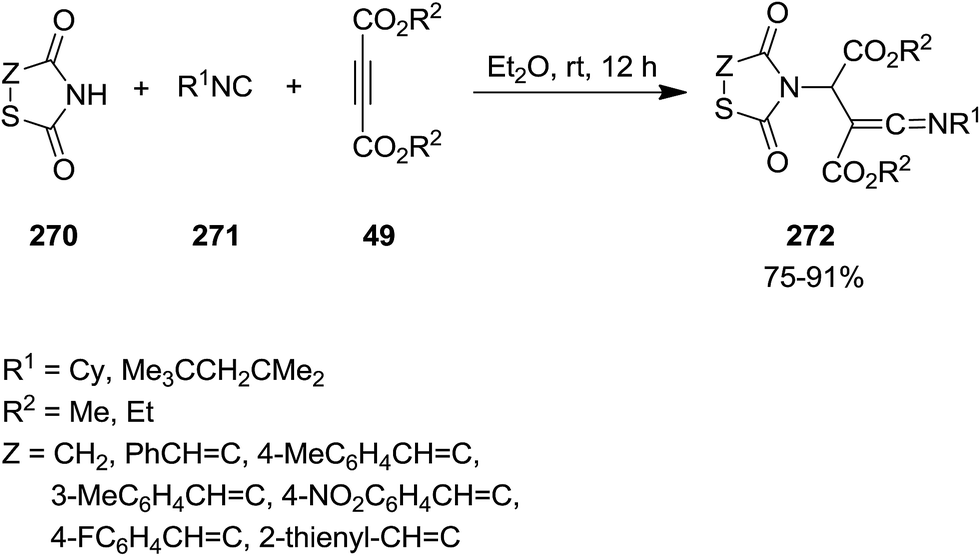
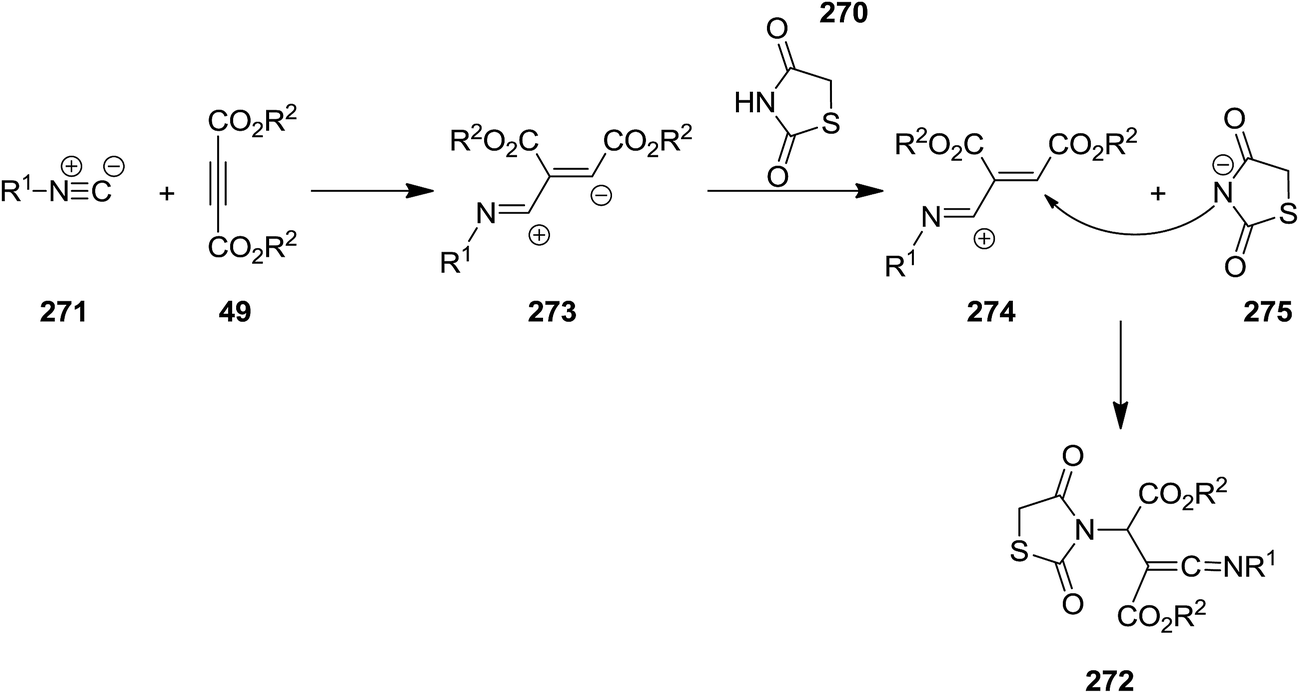
2.2.9 Thiazoles. Multi-substituted thiazolidine-2,4-dione derivatives 272 were synthesized from efficient and facile three-component reaction of thiazolidine-2,4-dione or 5-arylidene-2,4-thiazolidinediones 270, isocyanides 271 and acetylenedicarboxylates 49 under mild reaction conditions (Scheme 70).203 The obtained ketenimines which contained several functionalities could potentially be used for the synthesis of more complicated organic compounds. The plausible mechanism is depicted in Scheme 71. The mechanism involved generation of zwitterionic intermediate 273 followed by protonation and formation of 274 and 275. The product would be formed from attack of the resulting nucleophile on the positively charged 274.
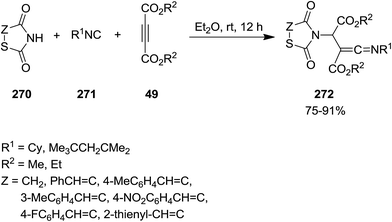 |
| | Scheme 70 | |
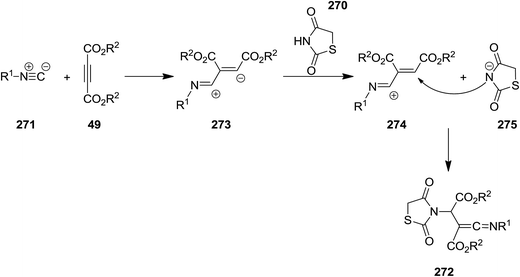 |
| | Scheme 71 | |
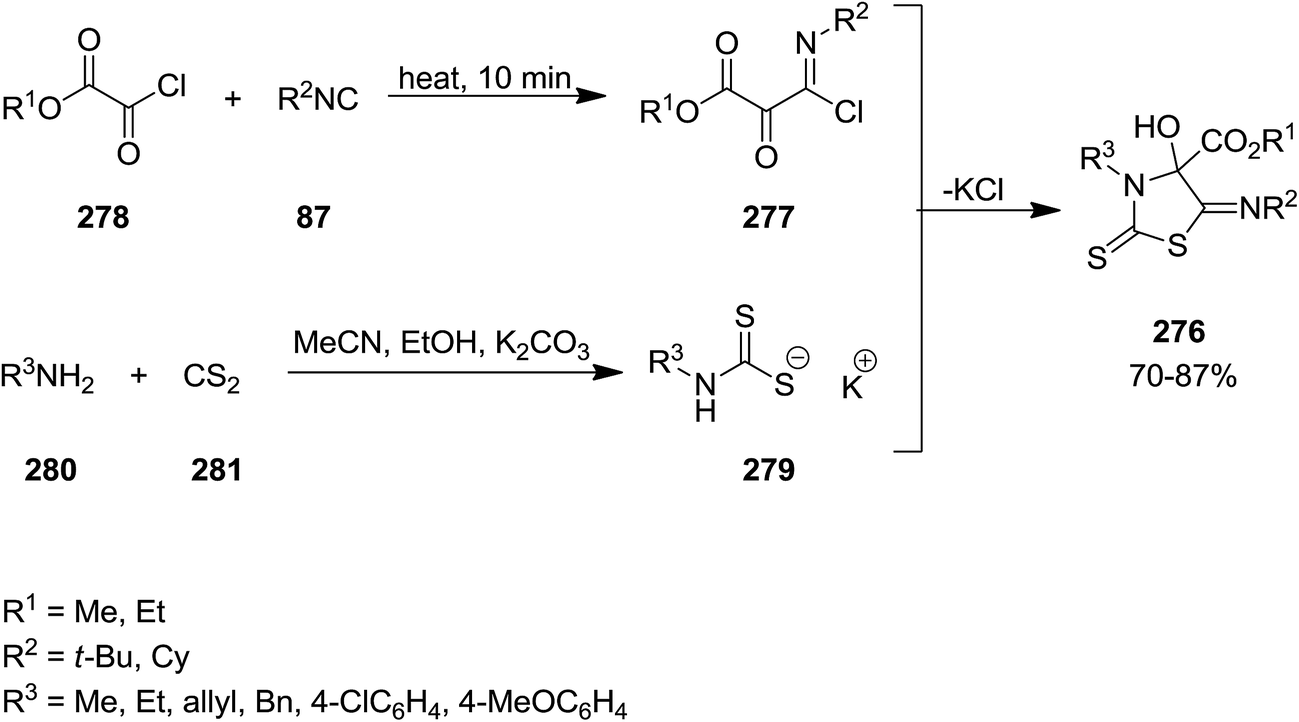
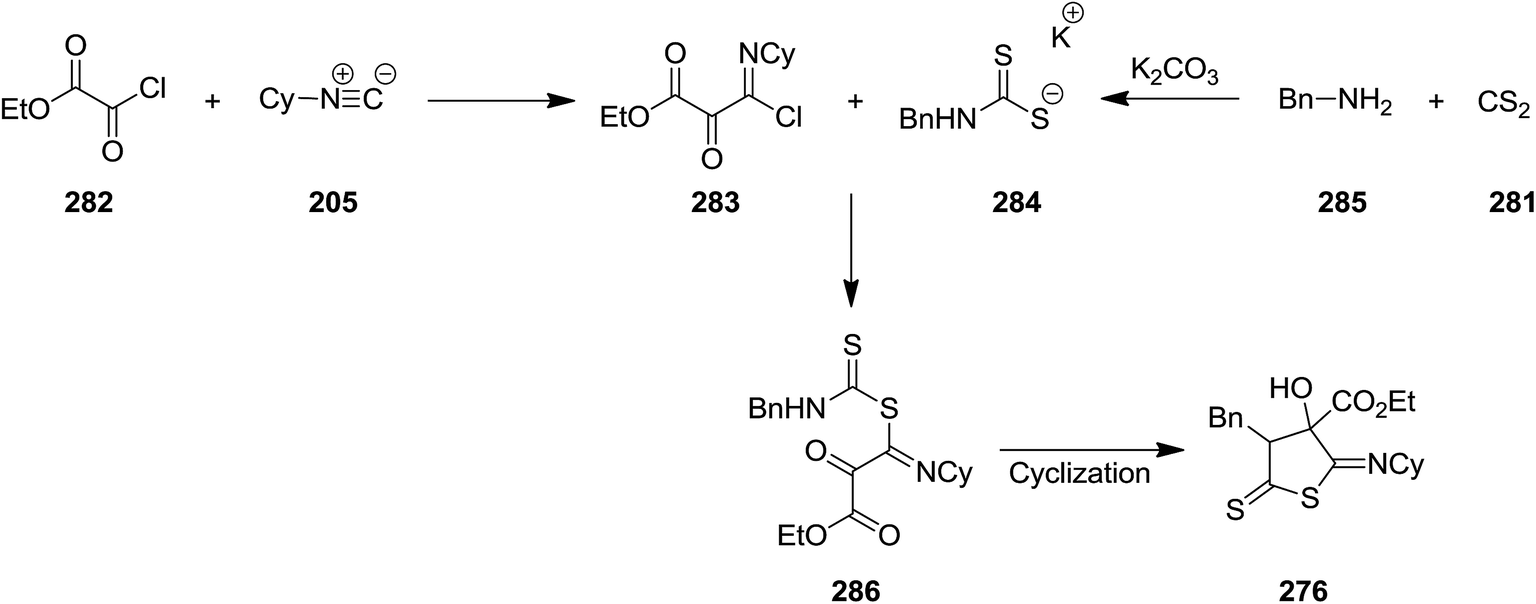
Yavari et al. developed base-promoted domino multi-component reaction to obtain 3-alkyl-5-(alkylimino)-4-hydroxy-2-thioxothiazolidine-4-carboxylate derivatives 276 (Scheme 72).204 In this synthetic strategy, the Nef-isocyanide adduct 277 derived from alkyl isocyanides 87 and alkyl chloroglyoxalates 278 and subsequently reacted with product 279 which was obtained from reaction of CS2 281 and amine derivatives 280 under mild reaction conditions in the presence of K2CO3 as a suitable base. The reaction preceded in short reaction time to afford the desired products in high yields. The reaction mechanism was based on formation of intermediate 283 followed by reaction with adduct 284 to afford a new intermediate 284. The later tolerated intramolecular cyclization to furnish the desired product (Scheme 73).
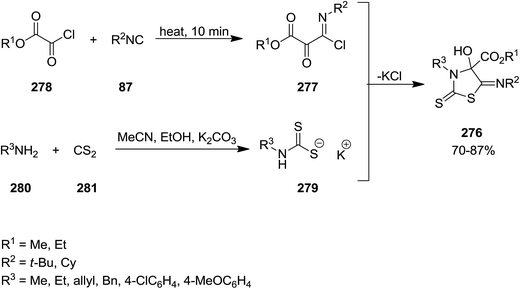 |
| | Scheme 72 | |
 |
| | Scheme 73 | |
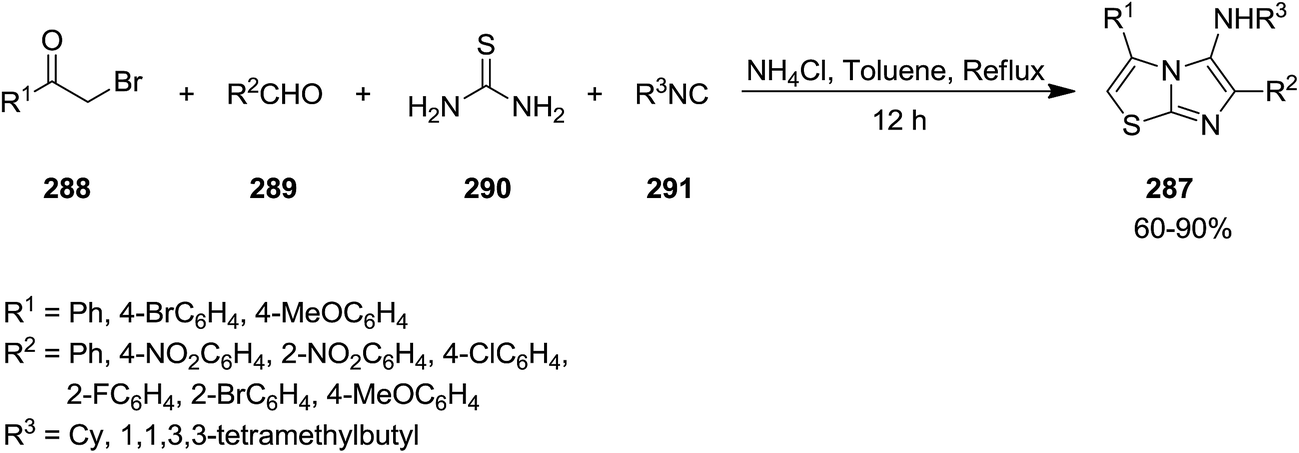
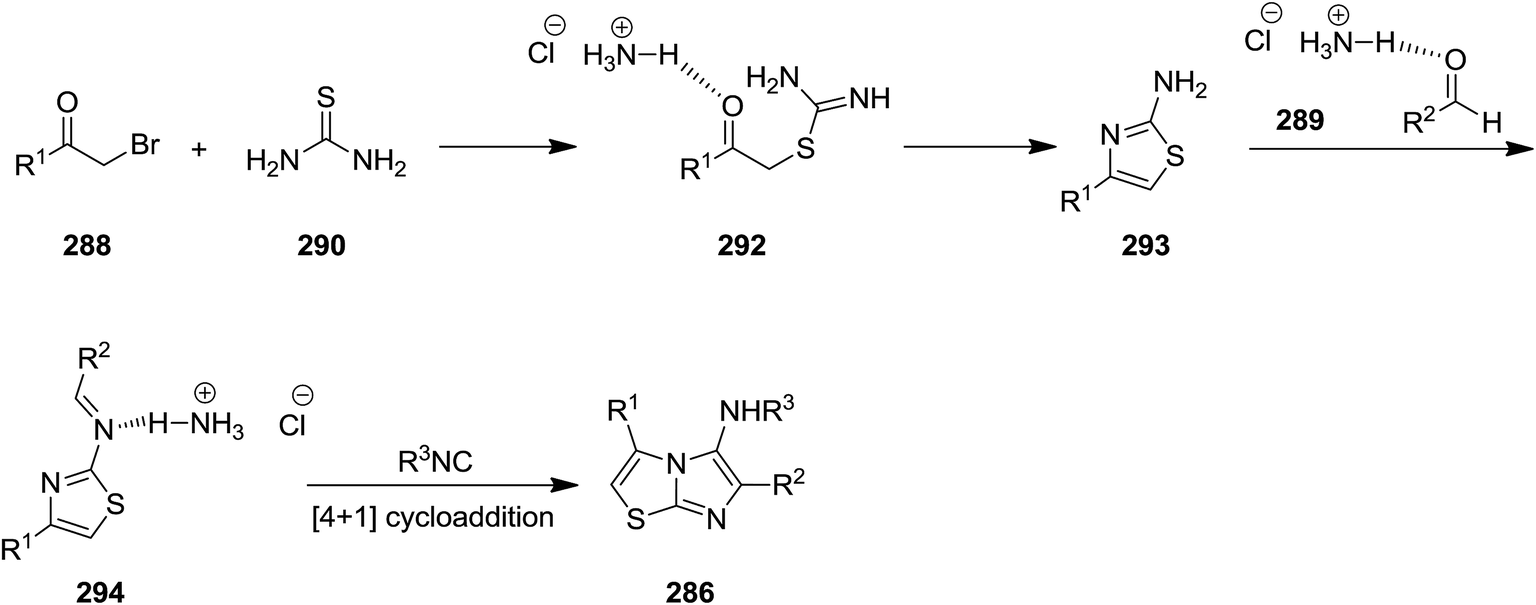
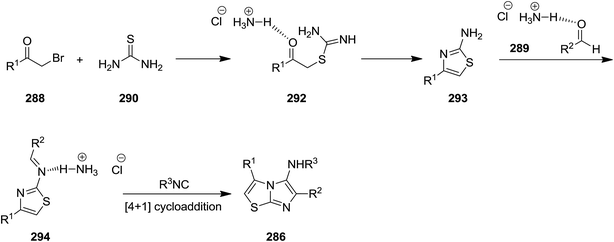
New derivatives of imidazo[2,1-b]thiazol-5-amine 287 were synthesized by using catalyst-free four component reaction of 2-bromoacetophenone derivatives 288, aromatic aldehydes 289, thiourea 290, and isocyanides 291 mediated by ammonium chloride (Scheme 74).205 High yields, mild reaction conditions and broad substrate scope were the advantages have been claimed for this synthetic protocol. It is worth noting that the presence of NH4Cl was essential for achieving high yields. The best results were obtained in the case of aldehydes with electron-withdrawing substituent at para position. The reaction mechanism was based on formation of intermediate 292 from 288 and 290 and subsequent formation of 293. The latter react with 288 to afford 294 which tolerated [4 + 1] cycloaddition to furnish the final product (Scheme 75).
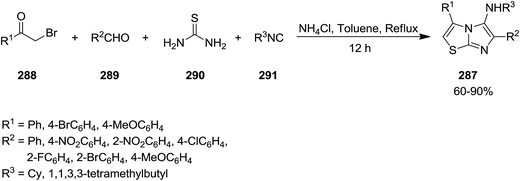 |
| | Scheme 74 | |
 |
| | Scheme 75 | |
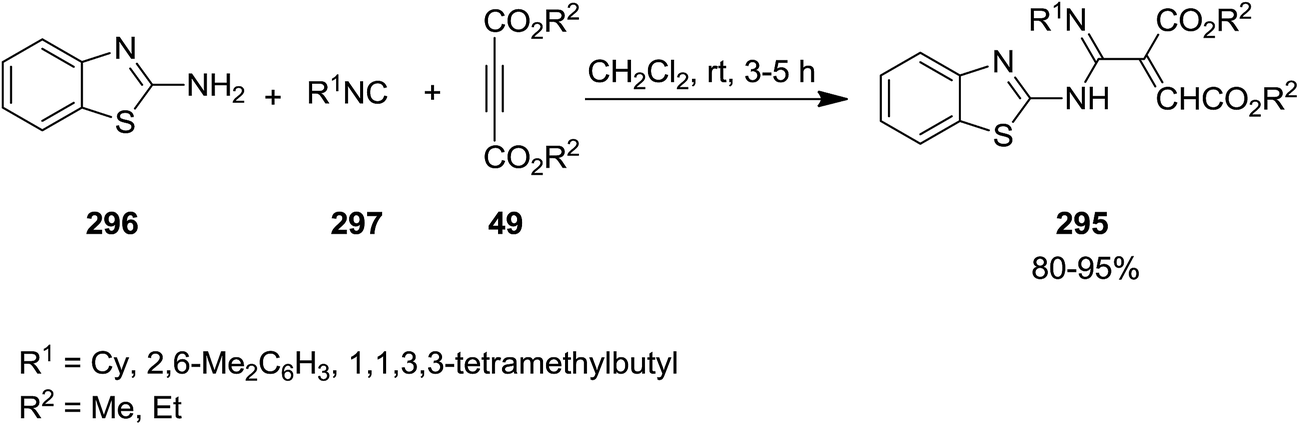
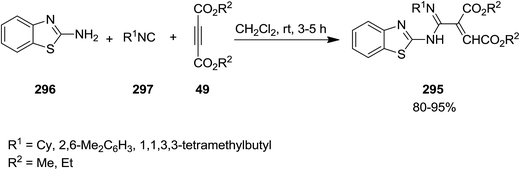
Substituted azadiene derivatives 295 were obtained in high yields via un-catalyzed three-competent reaction involving 2-aminobenzothiazole 296, isocyanides 297 and acetylenic esters 49 under mild reaction conditions (Scheme 76).206 The proposed mechanism involves the reaction of dialkyl acetylenedicarboxylates and isocyanide to afford 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 zwitterionic intermediate followed by its protonation. The latter was then attacked by 2-aminobenzothiazole to provide the desired product in high yields.
:1 zwitterionic intermediate followed by its protonation. The latter was then attacked by 2-aminobenzothiazole to provide the desired product in high yields.
 |
| | Scheme 76 | |
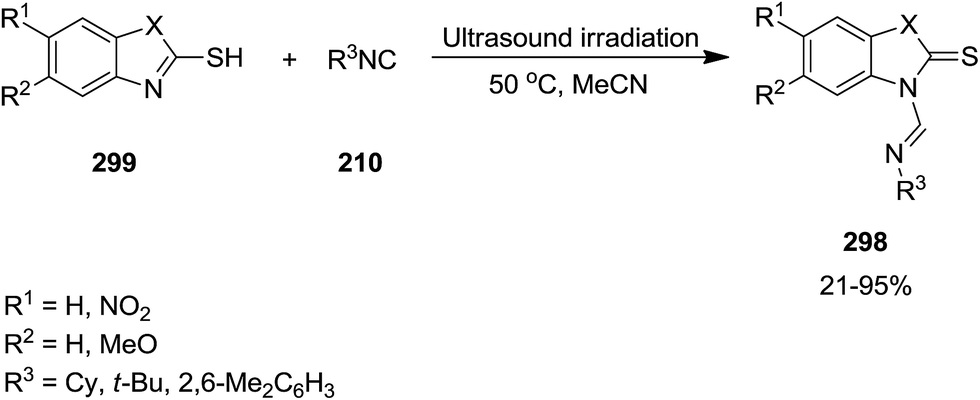
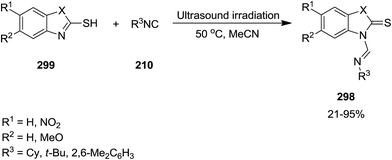
Green and ultrasonic-assisted process has been developed for the synthesis of formamidine scaffold 298 via reaction of 2-mercaptobenzothiazole and 2-mercaptobenzoxazole 299 with isocyanides 210 (Scheme 77). Despite the previously reported methods for the synthesis of formamidine framework which involved using large amount of metal-catalyst at high reaction temperature or inert atmosphere, this novel synthetic method led to the formation of the desired products in high yields under ambient atmosphere.207
 |
| | Scheme 77 | |
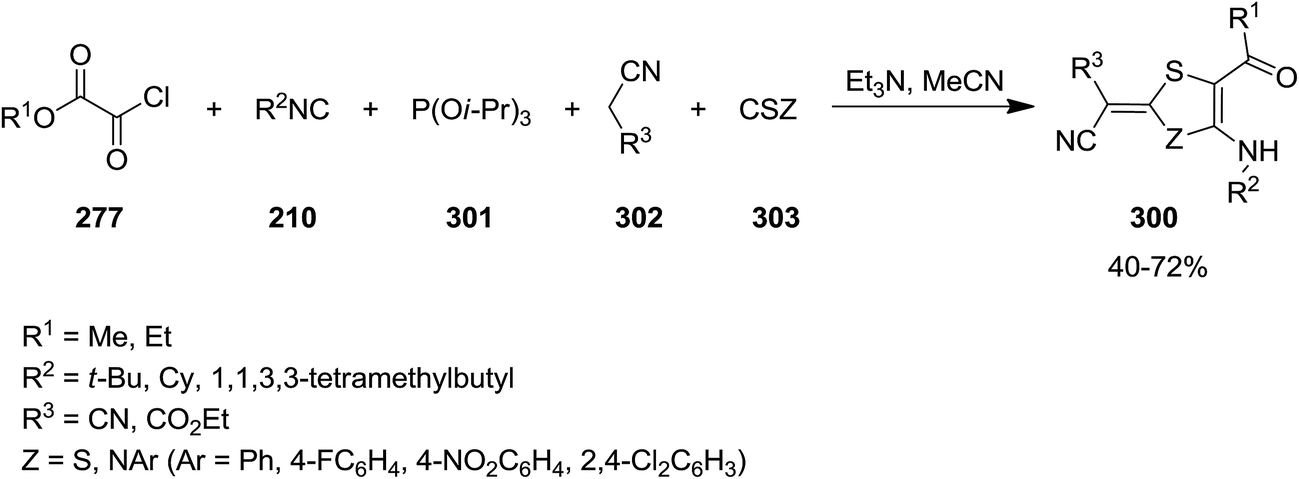
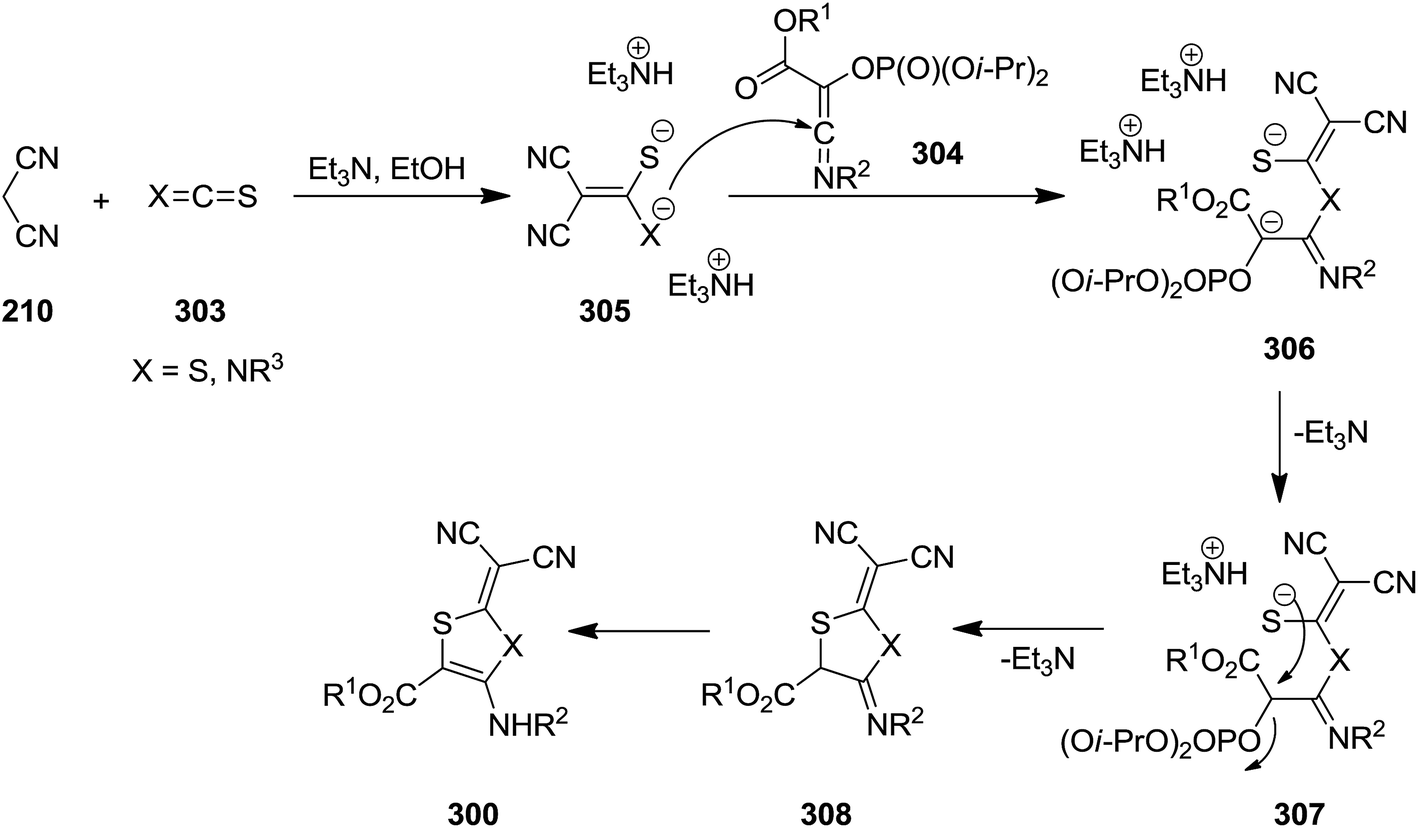
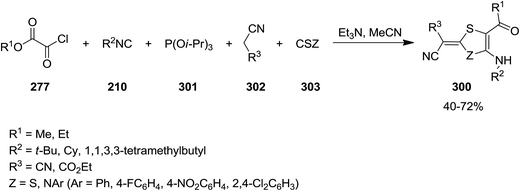
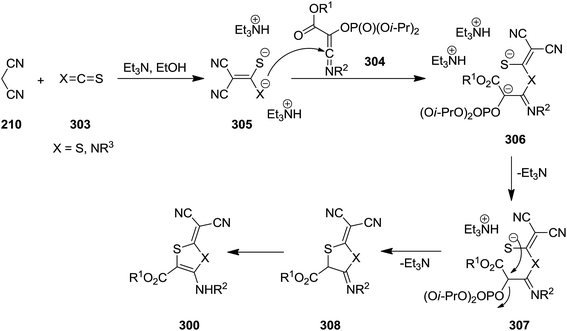
2,3-Dihydrothiazole-4-amino-5-carboxylate and 1,3-dithiole-5-amino-4-carboxylate 300 were synthesized via multicomponent reaction of triisopropyl phosphate 301, alkyl chloroglyoxalates 277, isocyanides 210, malononitrile or ethyl cyanoacetate 302 and isothiocyanate or carbon disulfide 303 (Scheme 78). The suggested reaction mechanism displayed in Scheme 79 involves the formation of an imidoyl chloride via Nef-isocyanide reaction followed by reaction with triisopropyl phosphate to generate ketenimines 304 in a Perkow-type reaction. The reaction of malononitrile or ethyl cyanoacetate 210 and isothiocyanate or carbon disulfide 303 led to adduct 305. This adduct in its triethylammonium salt form 306 reacted with ketenimine. The obtained intermediate 307 underwent proton transfer to afford a new intermediate which is subjected to phosphate elimination and intramolecular annulation to form 308, followed by tautomerization to give the final products 300 (Scheme 79).208
 |
| | Scheme 78 | |
 |
| | Scheme 79 | |
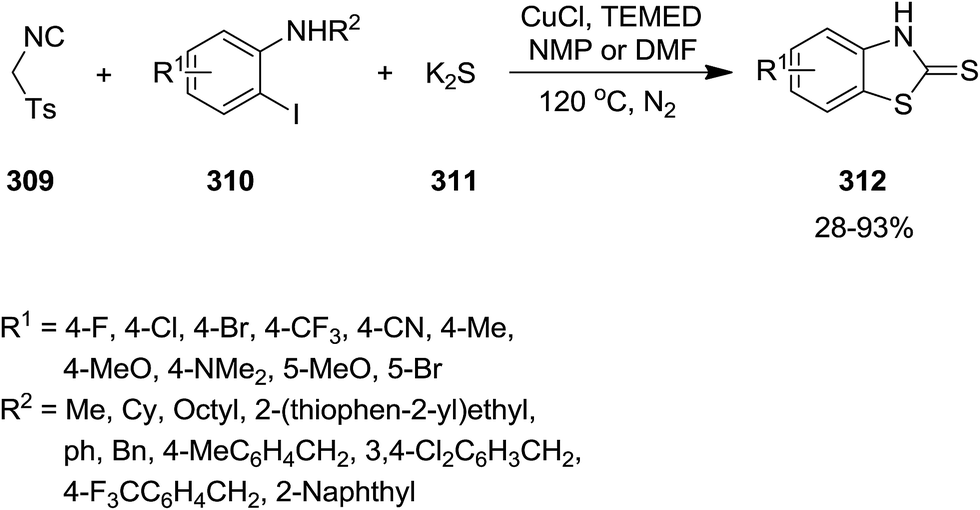
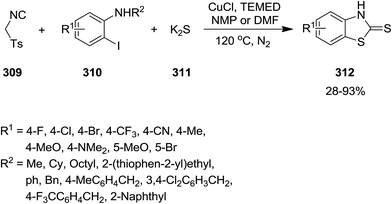
A diverse range of benzothiazolethiones 312 were synthesized efficiently from one-pot multi-component reaction of p-toluenesulfonylmethyl isocyanide 309, o-iodoaniline derivatives 310 and K2S 311 under copper catalysis in which isocyanide served as the source of C while K2S acted as source of S (Scheme 80).209 To investigate the generality of reaction, o-iodoanilines with various functional groups were examined. The results proved that the substrates with electron-donating groups resulted in superior yields. This observation was attributed to increase of electrophilicity of the amino group. It is worth noting that replacing o-iodoaniline with o-bromoaniline did not furnish the desired product in high yield.
 |
| | Scheme 80 | |
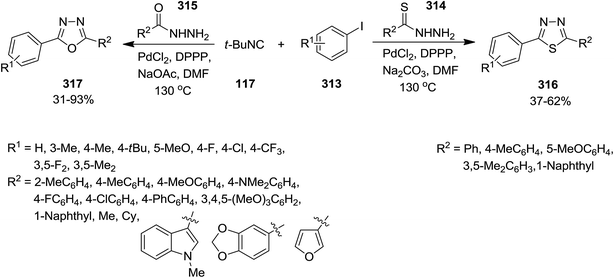
2.2.10 Diazoles. Using PdCl2 and DPPP as catalyst, Zhu et al. synthesized various diazole derivatives 316 and 317 in high to excellent yields through one-pot multi-component reaction of iodobenzenes 313, isocyanide 117 and hydrazide derivatives including benzothiohydrazide 314 and benzohydrazides 315 (Scheme 81).210 It is worth noting that the best choice of base in the case of benzohydrazides was NaOAc, while Na2CO3 was the most efficient base for reactions with benzothiohydrazide. The substrate scope was investigated by using various iodobenzenes containing electron-donating and electron-withdrawing functional groups and aromatic, aliphatic and heterocyclic benzohydrazides. Te results indicated the functional group tolerance. However, benzhydrazides with electron-rich substituents led to desired products in higher yields.
 |
| | Scheme 81 | |
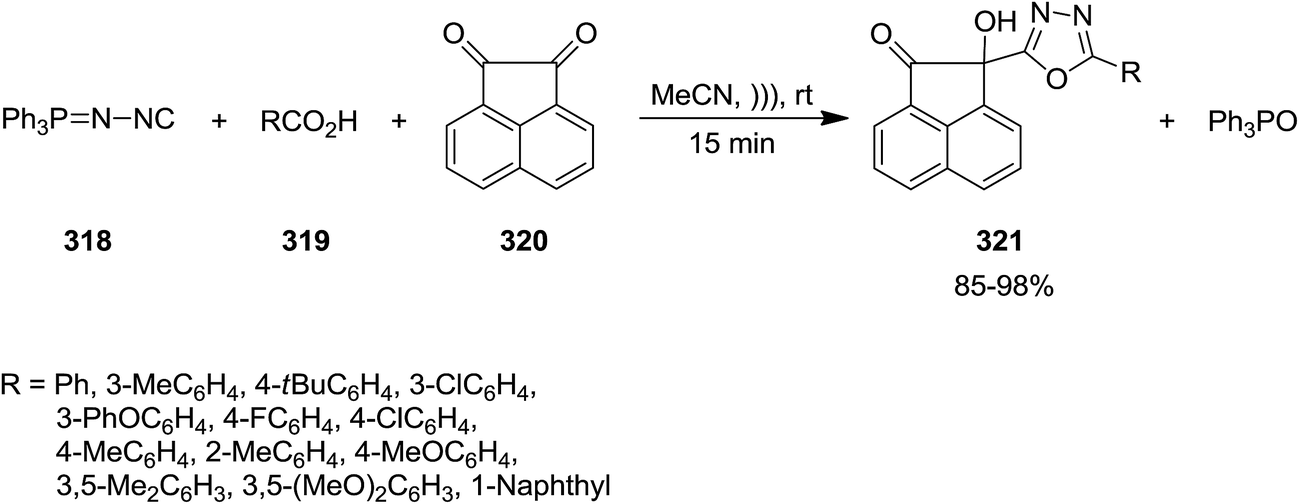
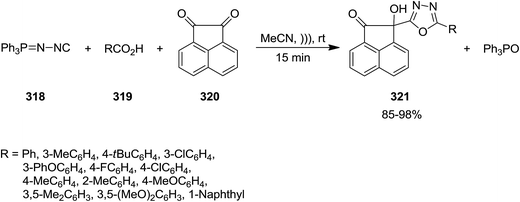
Ultrasonic-assisted synthesis of multi-substituted 1,3,4-oxadiazole derivatives 321 was reported by Ramazani and Joo et al. through multi-component reaction of (N-isocyanimino)triphenylphosphorane 318, aromatic carboxylic acids 319 and acenaphthoquinone 320 (Scheme 82).211 Ultrasonic irradiation not only played a dramatic role in reducing the reaction time, but also improved the yield of products. This novel protocol showed high functional group tolerance and could be applied for synthesis of various oxadiazoles.
 |
| | Scheme 82 | |
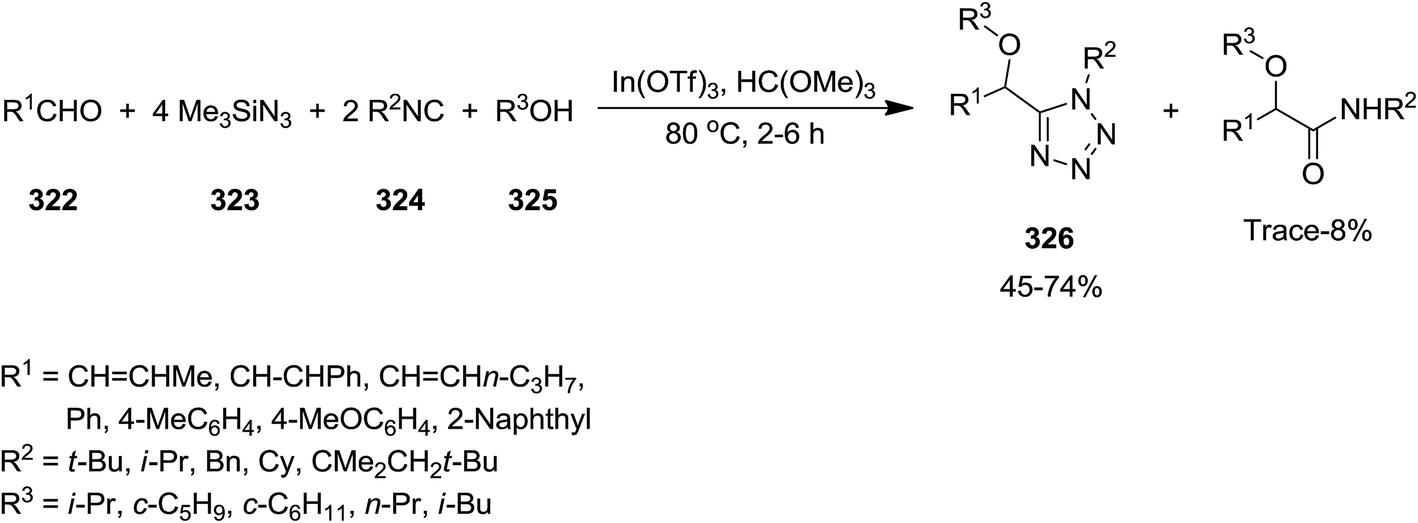
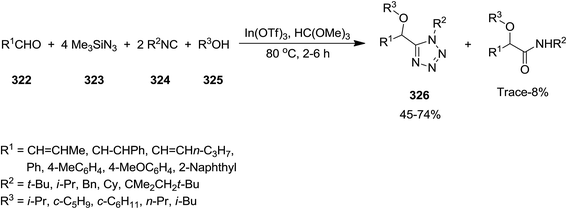
2.2.11 Tetrazole. In(III) Lewis acid catalysts, indium(III)triflate (In(OTf)3) and In(ONf)3 (Nf = n-C4F9SO2), were exploited to catalyze the synthesis of substituted 1H-tetrazole derivatives 326 via multi-component reaction of aldehydes 322, trimethylsilyl azide 323, isocyanides 324, and alcohols 325 without aliphatic moiety (Scheme 83). Regarding the mechanism of the reaction, it was proposed that the reaction of alcohol and aldehyde catalyzed by In(III) Lewis acid affords carboxonium. The obtained carboxonium undergoes isocyanide addition to generate nitrilium intermediate. Alcoholysis of trimethylsilyl azide and the formation of hydrazoic acid followed by its 1,3-dipolar cycloaddition to nitrilium resulted in the final product.212 Successful results by using cyclic mixed acetal 327 instead of aldehyde and formation of 328, confirmed that this reaction could be recognized as intramolecular types (Scheme 84).
 |
| | Scheme 83 | |
 |
| | Scheme 84 | |
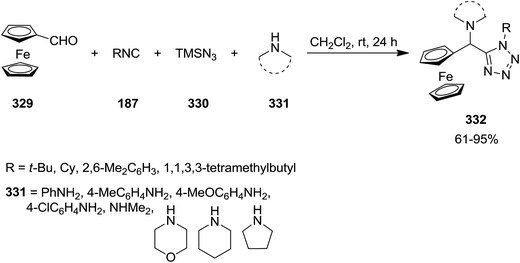
Bazgir et al. developed a novel catalyst-free synthetic strategy with wide substrate scope for the preparation of ferrocenyl dialkylamino tetrazoles and ferrocenyl arylamino tetrazoles 332. In this approach, the desired products were obtained in good to excellent yields via four-component reaction of ferrocenecarboxaldehyde 329, isocyanides 187, azidotrimethylsilane 330, and alkyl or aryl amines 331 under mild reaction conditions (Scheme 85).213 The reaction of aldehydes and amines led to the formation of the corresponding iminum ion which reacted with isocyanide to furnish an intermediate. Subsequent nucleophilic addition of the azide to this intermediate, followed by annulation resulted in the final product.
 |
| | Scheme 85 | |
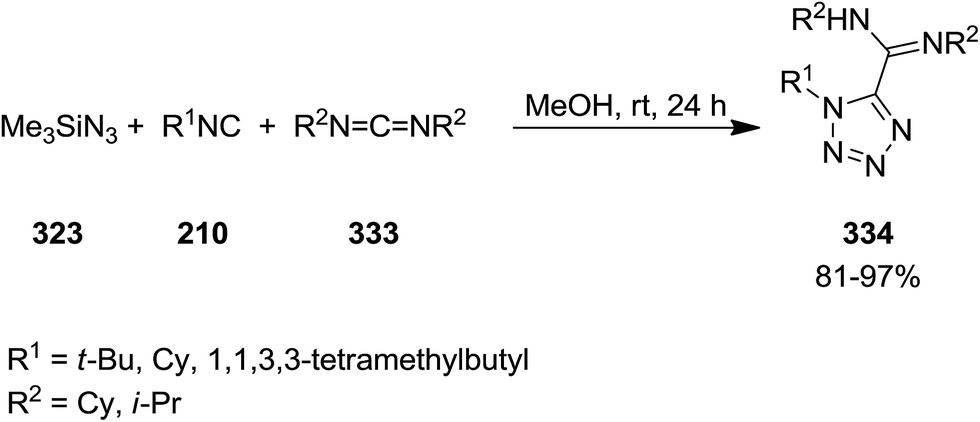
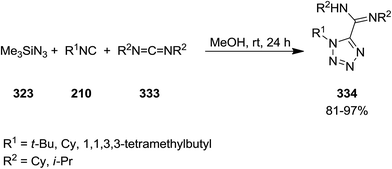
1,5-Disubstituted 1H-tetrazoles 334 were obtained via multi-component reaction of carbodiimides 333, isocyanides 210 and trimethylsilyl azide 323 in high yields (Scheme 86).214 The reaction proceeded at room temperature in methanol without the formation of any by-product. The simplicity of this procedure as well as high yields were the merits of this synthetic strategy.
 |
| | Scheme 86 | |
2.3. Six-member ring
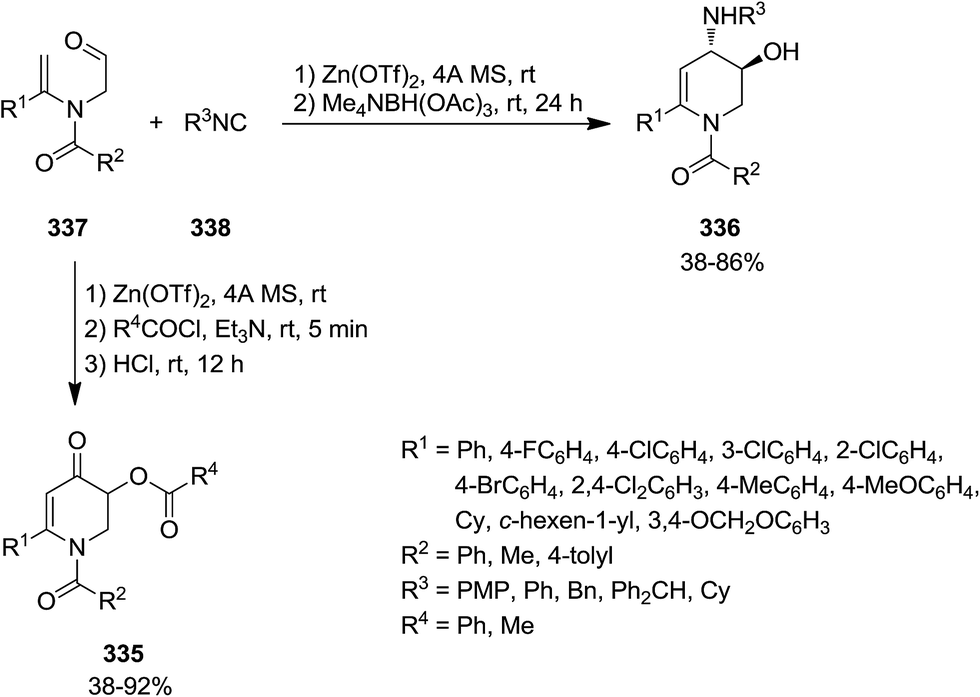
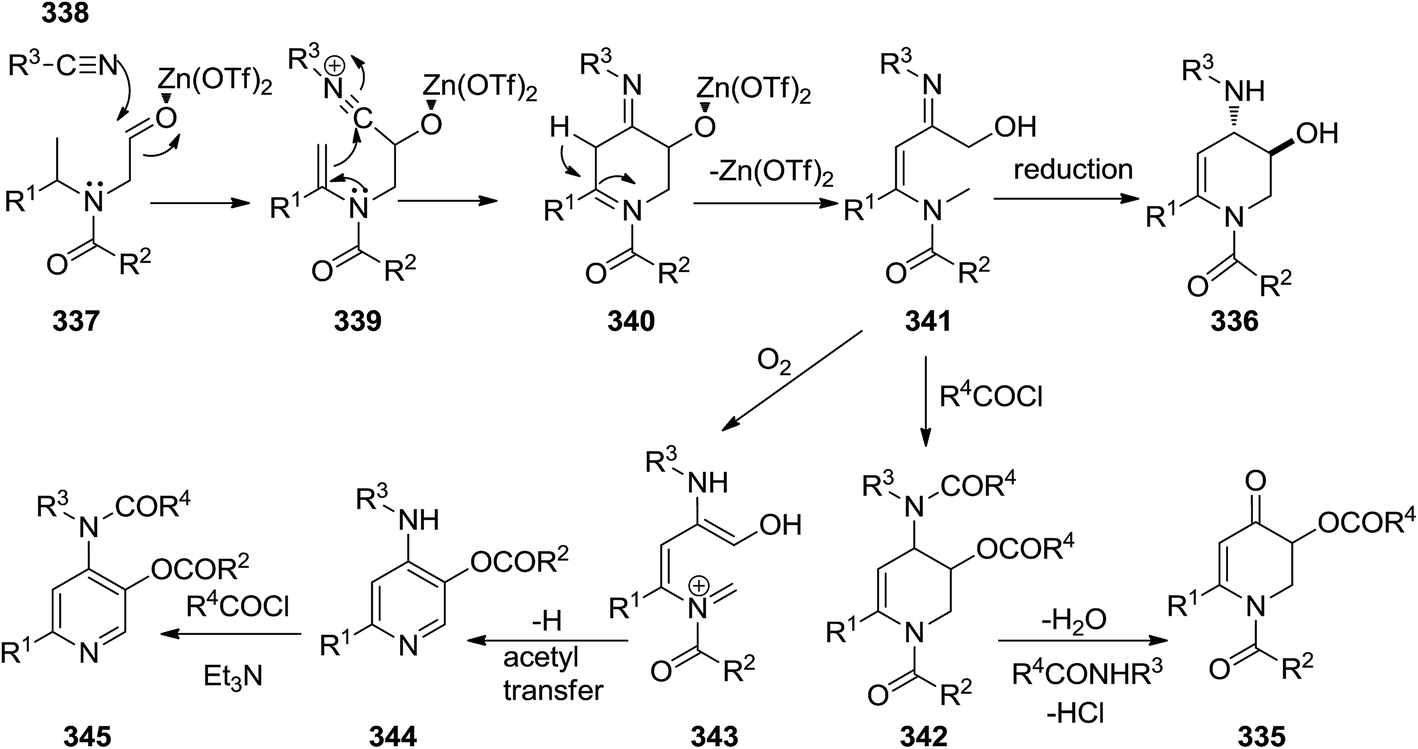
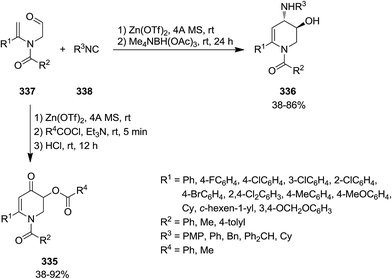
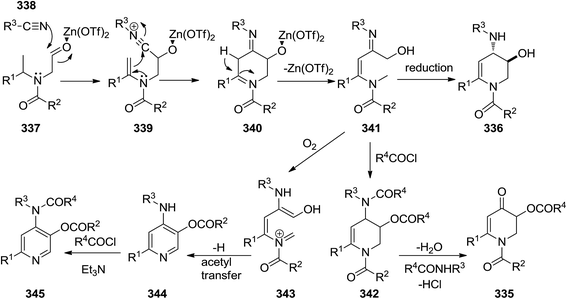
2.3.1 Pyridine. The synthesis of six-membered nitrogen-containing heterocycles including, poly-substituted 2,3-dihydropyridin-4(1H)-one 335, 1,2,3,4-tetrahydropyridine 336, and pyridine derivatives, was reported through cascade reaction (Scheme 87). The tandem reaction commenced by [5 + 1] cycloaddition of N-formylmethyl substituted tertiary enamides 337 to isocyanides 338 in the presence of Zn(OTf)2 as Lewis acid to yield 339–341.215 The subsequent reduction of intermediate with Me4NBH(OAc)3 resulted in 1,6-disubstituted trans-3-hydroxy-4-arylamino- or 1,2,3,4-tetrahydropyridine 336 while acylation led to 342 and subsequent hydrolysis resulted in 1,6-disubstituted 3-acyloxy-2,3-dihydropyridin-4(1H)-ones 343. 3-Acyloxy-4-aminopyridines 344 were obtained by mild oxidation and aromatization and acyl group transfer yielded 345 (Scheme 88).
 |
| | Scheme 87 | |
 |
| | Scheme 88 | |
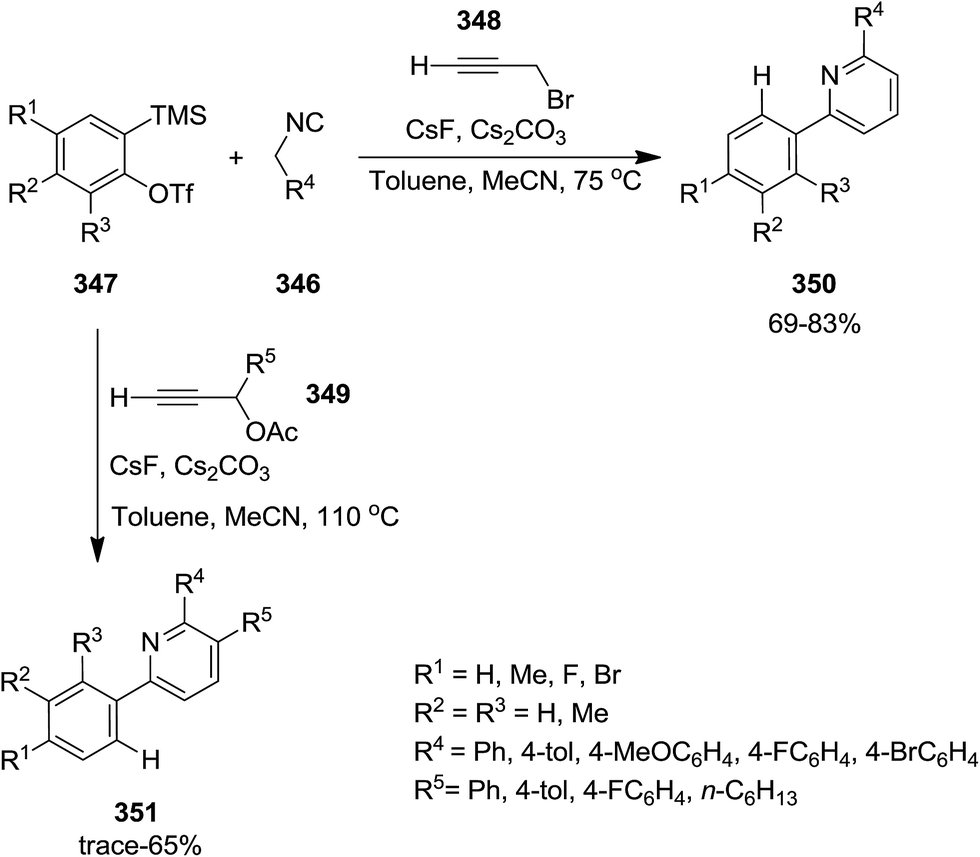
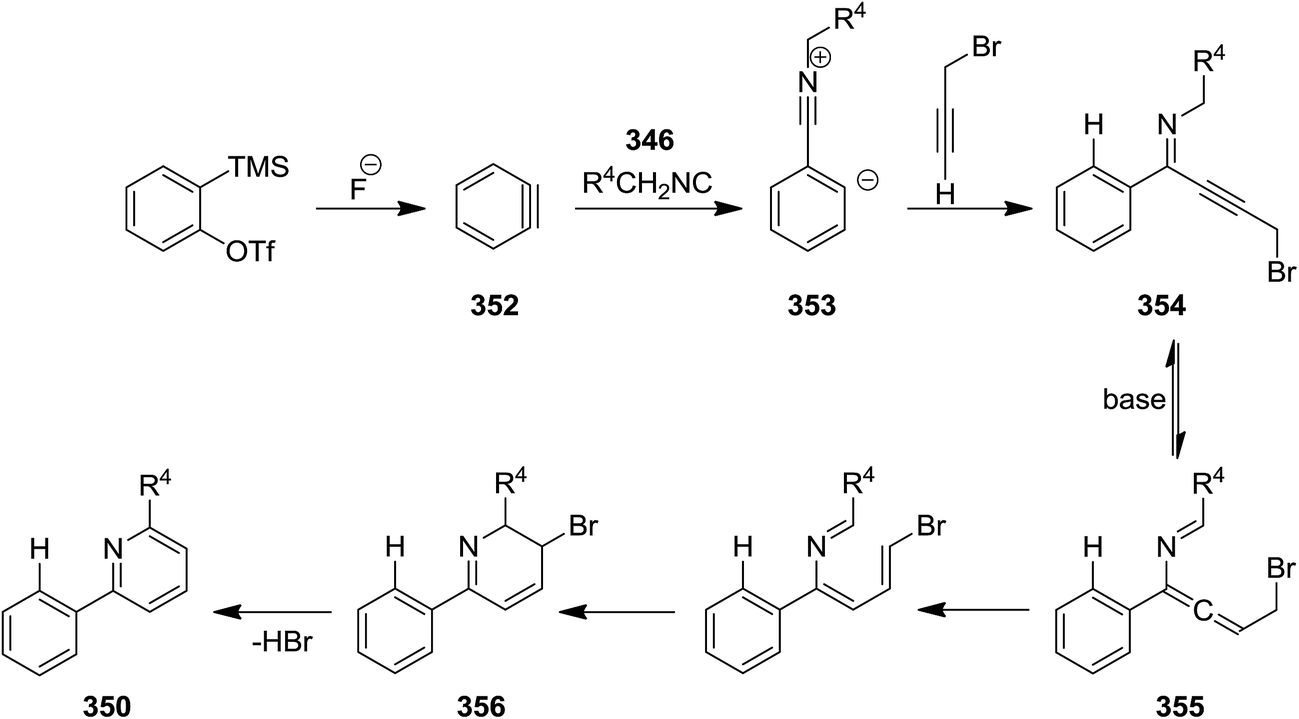
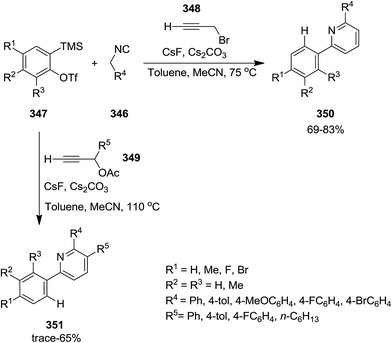
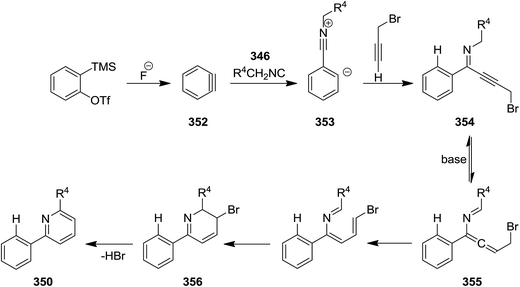
Three-component reaction of isocyanides 346, in situ formed from arynes 347, and 3-bromo- or 3-acetoxypropynes 348 and 349 furnished di- and tri-substituted pyridine derivatives 350 and 351. To confirm the generality of the reaction various isocyanides including electron-deficient and electron-rich ones were tested (Scheme 89). The proposed mechanism was based on in situ formation of aryne 352 which was attacked by isocyanide 346 to generate zwitterionic intermediate 353. Subsequent entrapment of this intermediate by alkyne led to the formation of N-alkynyl imine species 354 which tolerated 1,5-H shift to produce allenyl imine intermediate 355. 1,3-H shift resulted in azatrienes (1Z)- and (1E)-species 356. 6π-Electrocyclic reaction followed by HBr elimination afforded the desired products (Scheme 90). The simplicity of procedure, obtaining high yields and regioselectivity were the merits of this synthetic protocol.216
 |
| | Scheme 89 | |
 |
| | Scheme 90 | |
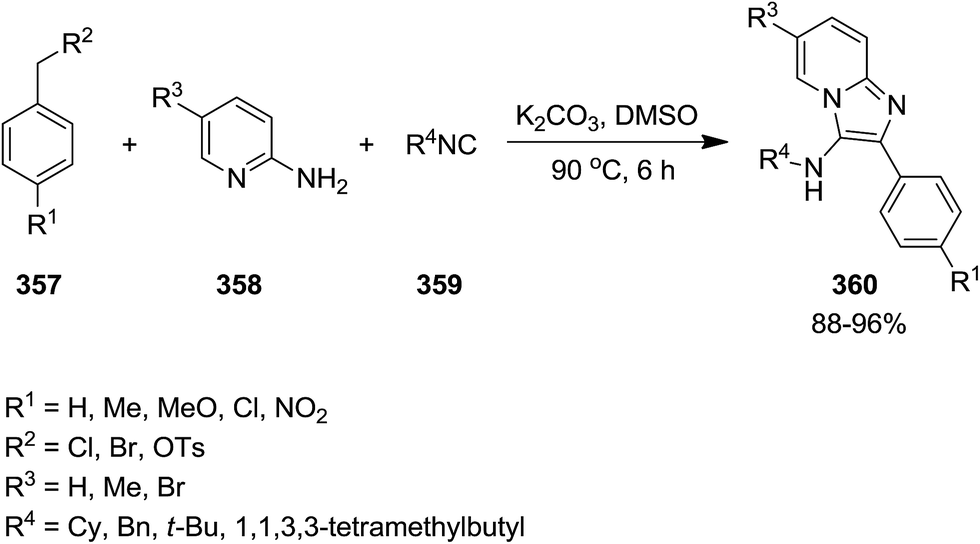
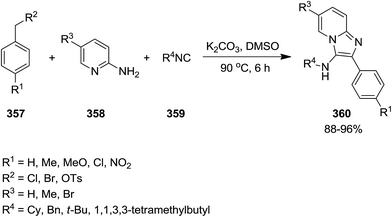
Adib et al. developed an efficient one-pot synthetic protocol with wide substrate scope for the synthesis of imidazo[1,2-a]pyridine derivatives 360 from reaction of benzyl halides or benzyl tosylates 357, 2-aminopyridines 358 and isocyanides 359 (Scheme 91).217 It was suggested that initial oxidation of benzyl halides or benzyl tosylates affords the corresponding aldehydes which tolerated three-component reaction with other reagents to furnish the final product in high yields.
 |
| | Scheme 91 | |
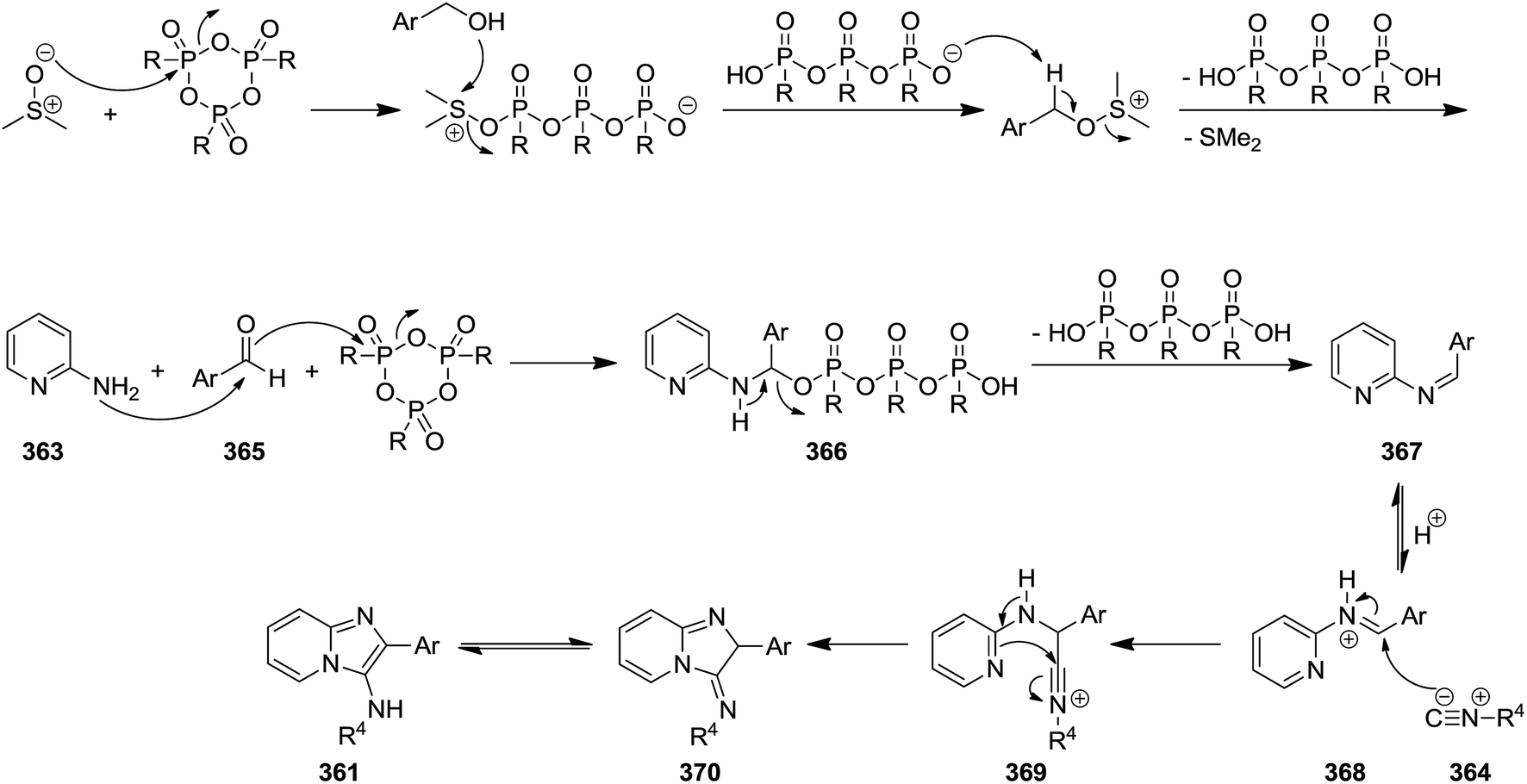
Propylphosphonic anhydride/DMSO (®T3P)-promoted one-pot synthesis of imidazo[1,2-a]pyridine derivatives 361 was achieved via multi-component reaction of 2-aminopyridines 362, alcohols 363 and isocyanides 364 (Scheme 92).218 This process involved in situ oxidation of alcohol to aldehyde 365 through formation of 366 and 367 species and its reaction with other components to form intermediates 368–370. ®T3P served as an activator and Schiff base (Scheme 93). Beside high yields, mild reaction condition and simplicity as well as cascade nature can be considered as the advantages of this synthetic process.
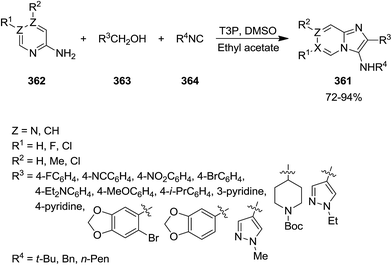 |
| | Scheme 92 | |
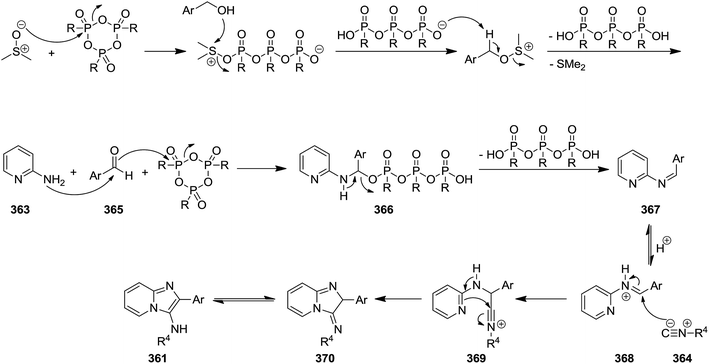 |
| | Scheme 93 | |
To furnish a solution to the practical difficulties of isocyanides, Guchhait et al. developed a one-pot protocol for the straightforward formation of isocyanide 371 via formylation of amine 372 and dehydration of formamide 375 and its subsequent use in conventional isocyanide multi-component reactions such as Groebke–Blackburn–Bienayme (GBB) reaction (reaction with azines 373 and aldehydes 374) (Schemes 94 and 95).219 It was found that the entity of base and dehydrating agent and their amounts played important roles in this procedure. p-Toluene sulfonyl chloride and DABCO were recognized as the most efficient dehydration agent and base respectively. Furthermore, altering the quantities of these two reagents influenced the yield of reaction remarkably. Acetonitrile was found the choice of solvent for this synthetic procedure.
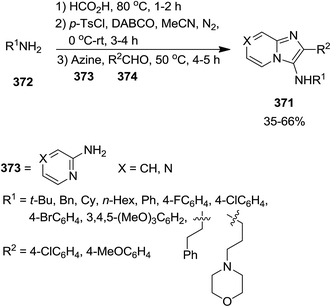 |
| | Scheme 94 | |
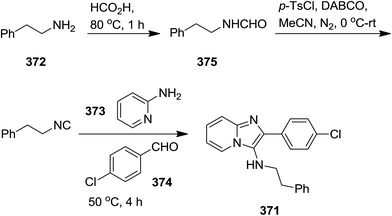 |
| | Scheme 95 | |
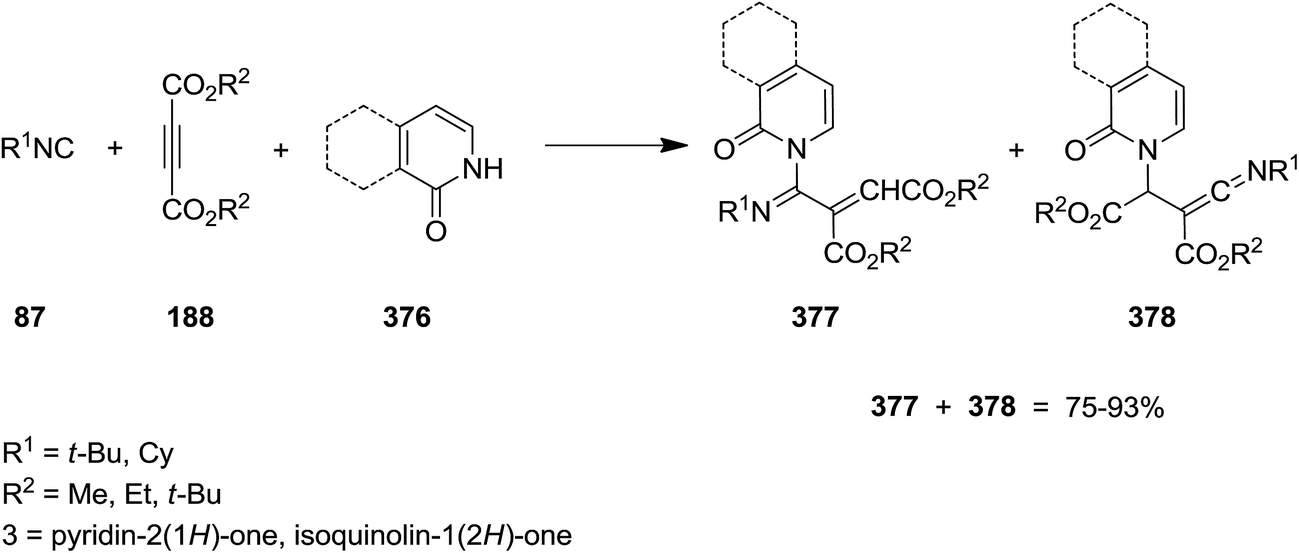
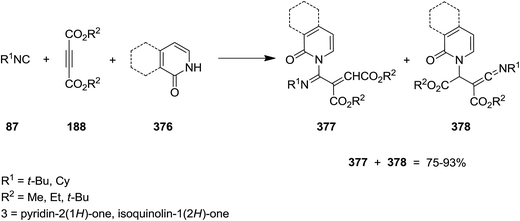
Yavari and his co-workers disclosed a novel method for preparation of poly-functionalized 1-azabuta-1,3-dienes 377 (Scheme 96).220 The insertion of isocyanide 87 to dialkyl acetylenedicarboxylate 188 led to the formation of an 1:1 intermediate which subsequently could be trapped by pyridin-2(1H)-one and isoquinolin-1(2H)-one 376 to afford 1-azabuta-1,3-dienes 377 as the main product. In some cases, ketenimines 378 were also obtained as minor products.
 |
| | Scheme 96 | |
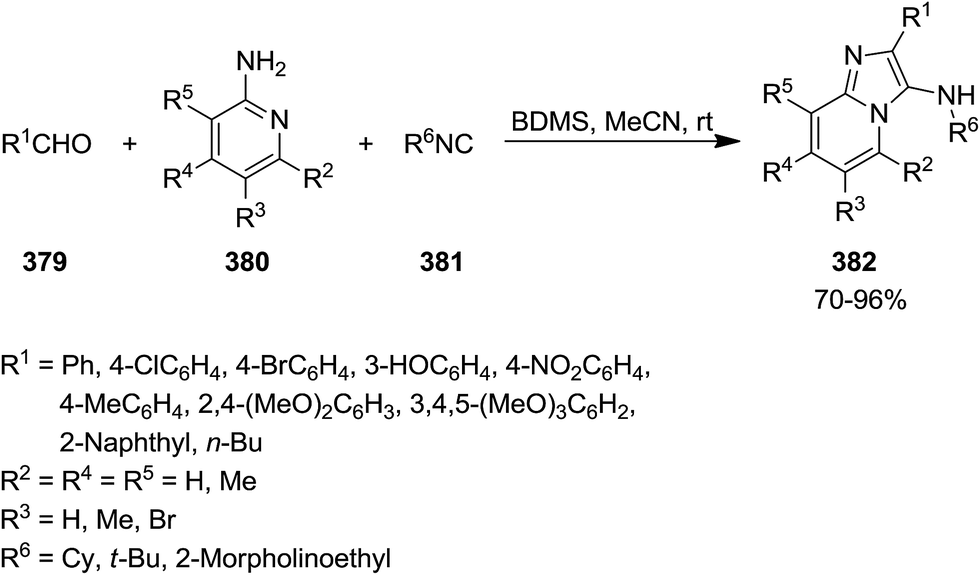
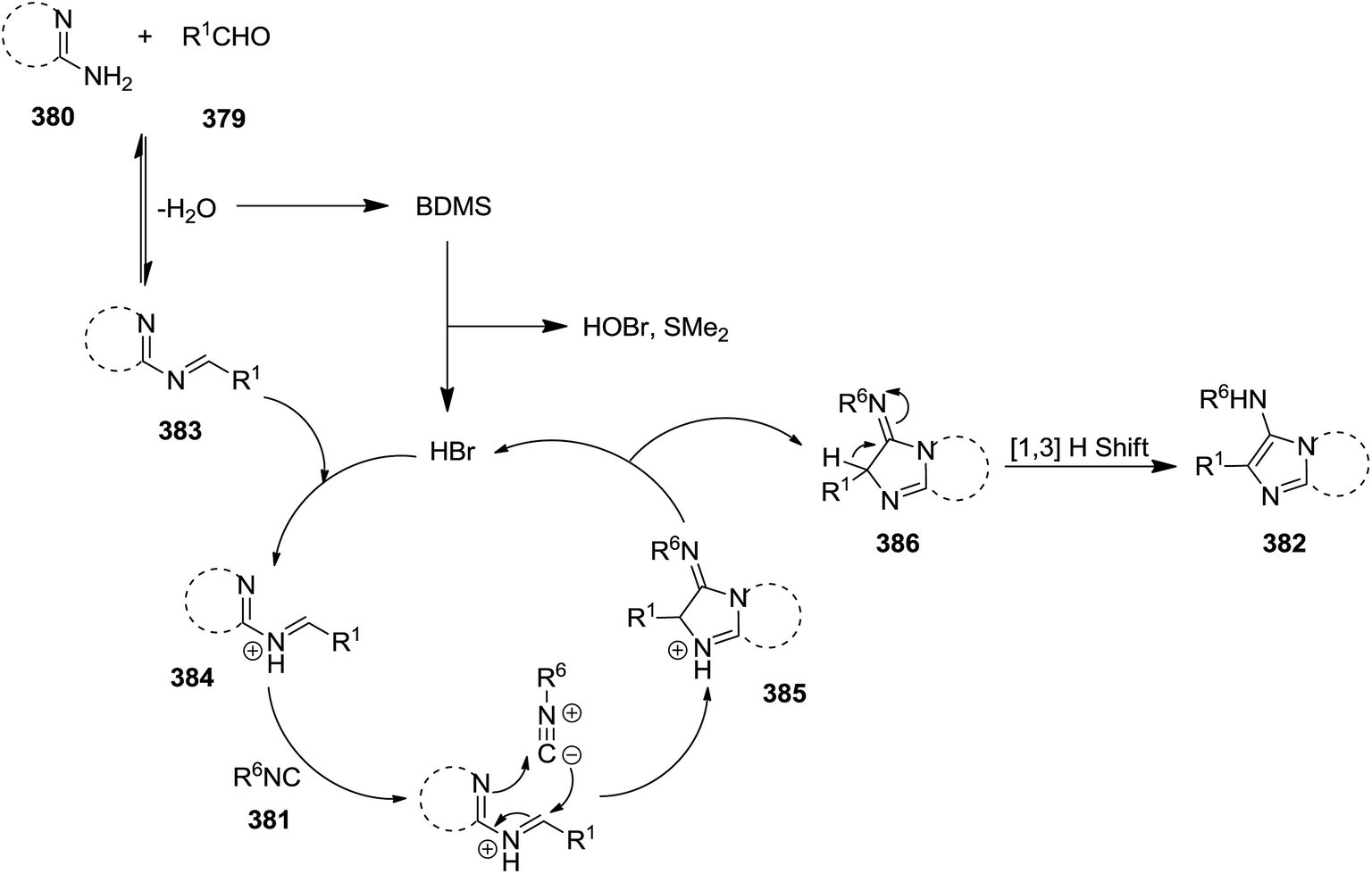
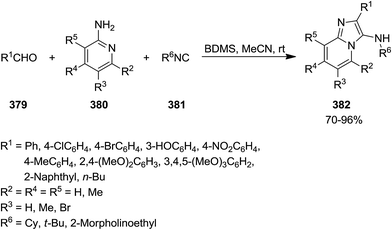
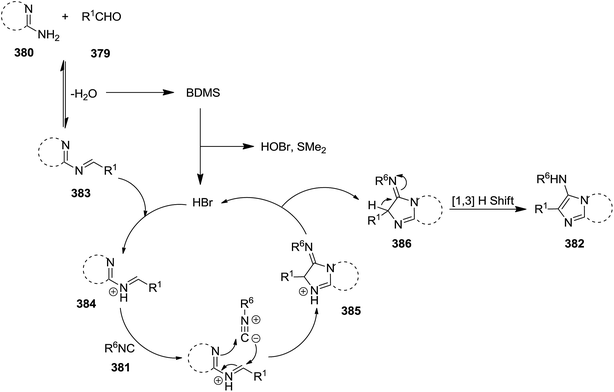
Bromodimethylsulfonium bromide (BDMS) was used as catalyst to promote Ugi-reaction of divergent aromatic aldehydes 379, aromatic amidinesin 380, and isocyanides 381 under mild reaction conditions (Scheme 97).221 This synthetic procedure resulted in the formation of a series of imidazo[1,2-a]pyridine derivatives 382 with potential utility in fluorescent probe for biomedical and clinical applications, mainly for diagnosis. Low catalyst loading (5 mol%), high yields, short reaction time (0.5–2.5 h), simplicity and broad substrate scope were the claimed advantages of this synthetic protocol. The plausible mechanism involved the reaction of an appropriate amidine and aldehyde resulted in the formation of imine 383 and H2O. The reaction of water with catalyst generated SMe2, HOBr, and HBr. Protonation of imine with HBr resulted in the formation of an intermediate 384 which underwent [4 + 1] cycloaddition reaction with an appropriate isocyanide to afford a new intermediate 385. The 1,3-hydrogen shift of the latter 386 led to the formation of the final product (Scheme 98).
 |
| | Scheme 97 | |
 |
| | Scheme 98 | |
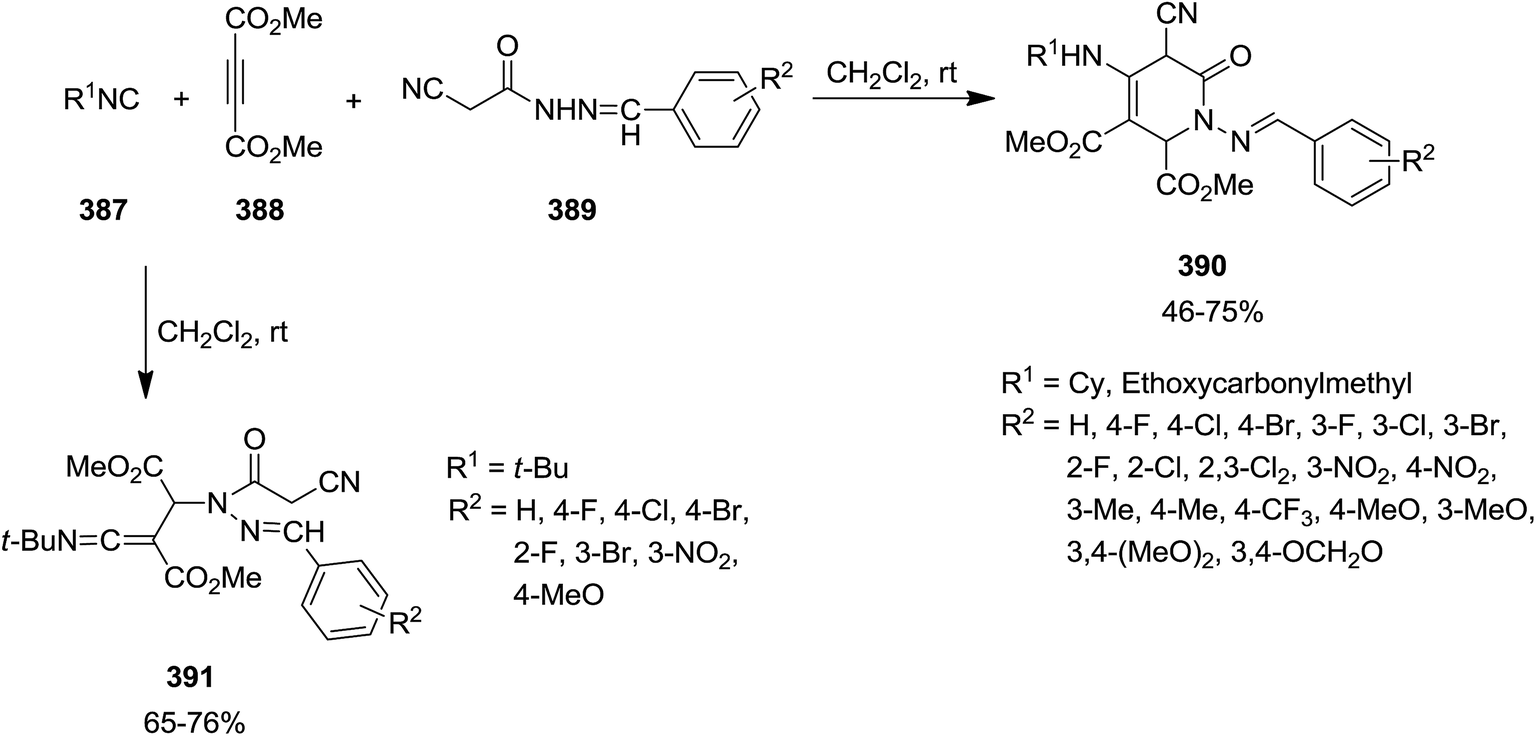
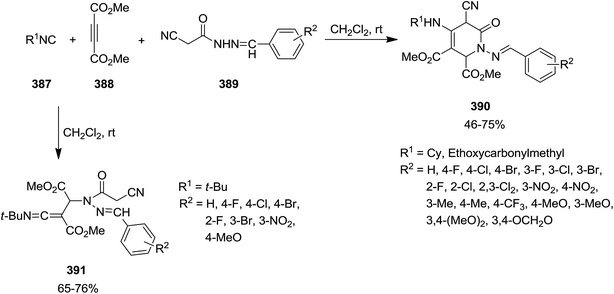
Li, Wen et al. exploited catalyst and additive-free multi-component reaction of isocyanides 387, dimethyl acetylenedicarboxylate 388, and N-arylidene-2-cyanoacetohydrazides 389 for synthesis of a series of pyridin-2-ones 390 in a regioselective manner (Scheme 99).222 It is worth mentioning that intramolecular nucleophilic attack did not occur by using tert-butyl isocyanide and produced compound 391. Moreover, the solvent dramatically affected the reaction and it was observed that the reaction did not proceed in EtOH. Simplicity of procedure, high yields and using readily available reagents were the merits of this process.
 |
| | Scheme 99 | |
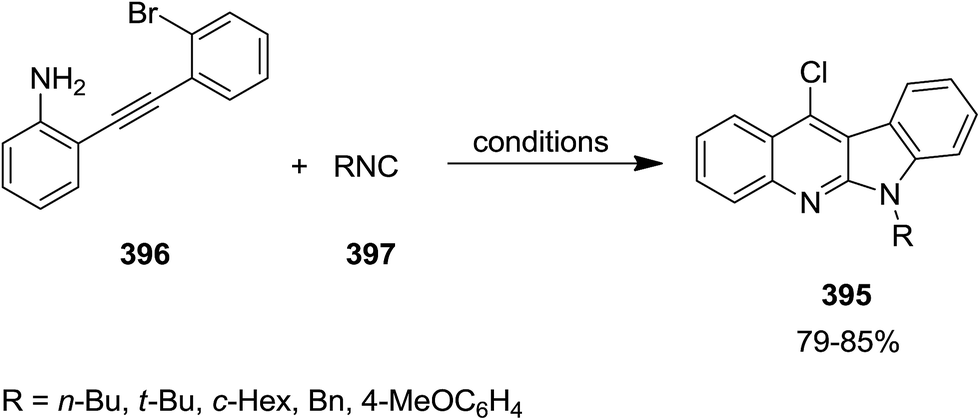
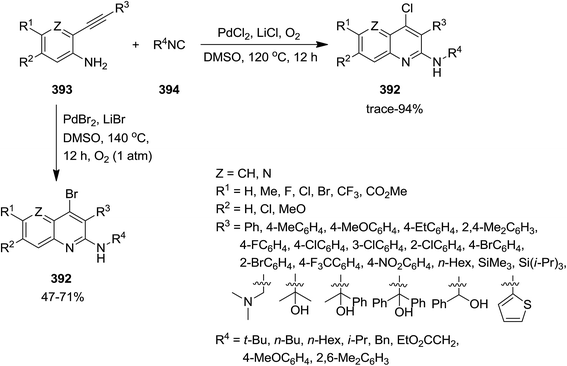
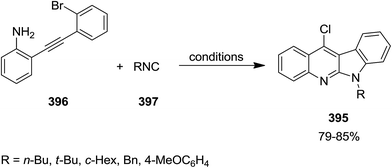
2.3.2 Quinolines. Jiang et al. developed a general and facile Pd-catalyzed approach for an efficient synthesis of various 4-halo-2-aminoquinolines 392. This strategy consisted of aerobic oxidative intermolecular cyclization of 2-ethynylanilines 393 with isocyanides 394 (Scheme 100). The generality of this reaction was studied using various isocyanides and differently substituted 2-ethynylanilines. Notably, it was found that the yields of 4-bromo-2-aminoquinolines were lower than 4-chloro-2-aminoquinolines. This approach was also employed for the synthesis of 6H-indolo[2,3-b]quinolines 395 via an intramolecular Buchwald–Hartwig cross-coupling of 396 and isocyanide 397 in two-steps but in one-pot fashion (Scheme 101).223
 |
| | Scheme 100 | |
 |
| | Scheme 101 | |
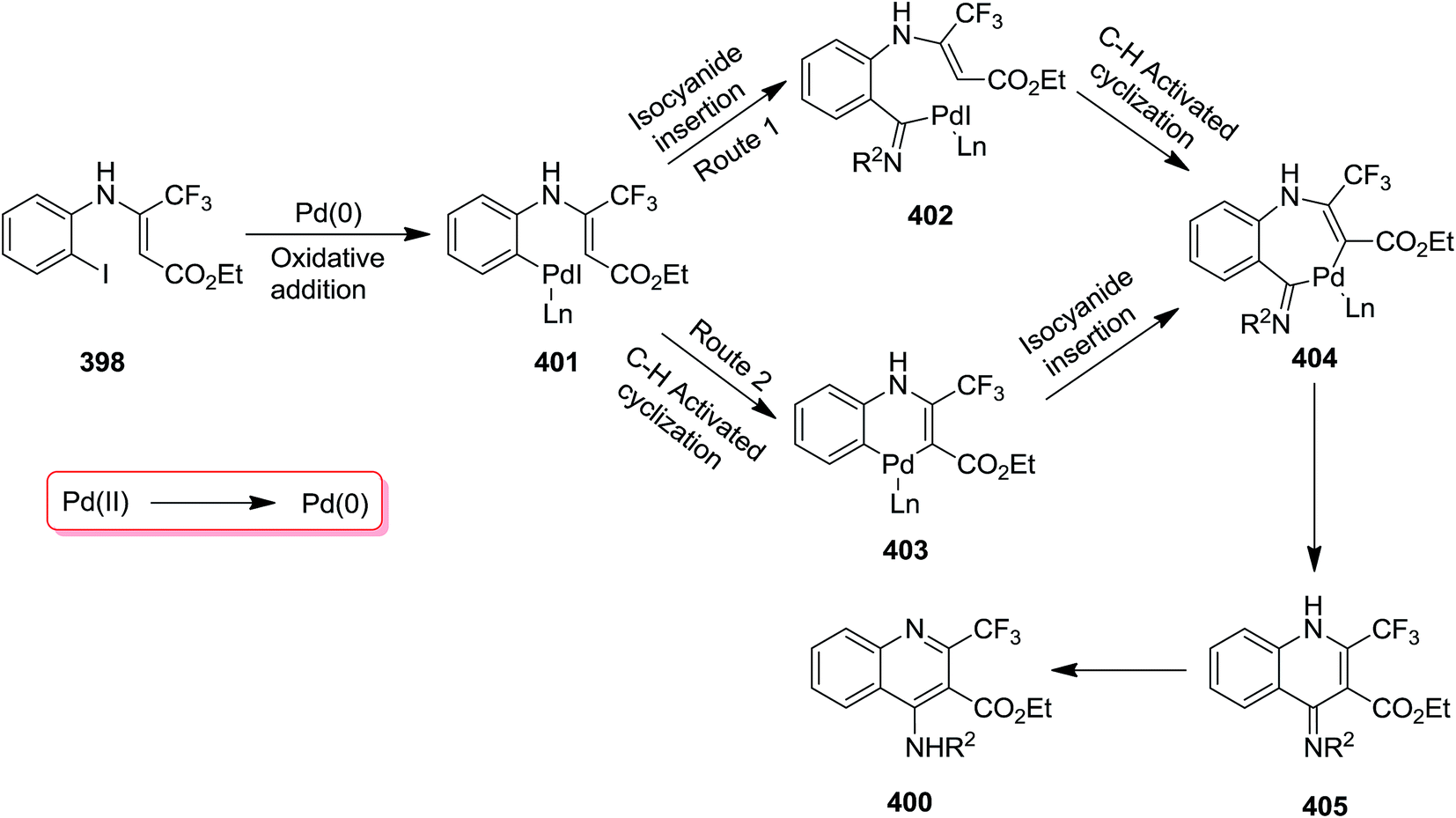
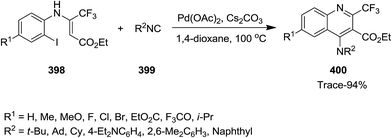
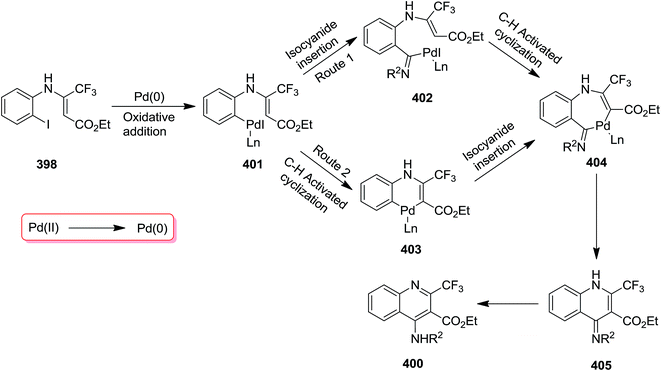
Using Pd as catalyst, tandem reaction of N-(ortho-iodo)aryl enamines 398 and isocyanides 399 was performed under mild reaction conditions to afford bioactive 4-amino-2-trifluormethyl quinoline 400 in moderate to high yields (Scheme 102).224 Cs2CO3 was found as the most efficient base for this reaction. The effect of atmosphere under which the reaction took place was studied by performing the reaction under N2 and O2. The results implied that the reaction was not sensitive to atmosphere and almost similar yields were obtained under both conditions. The proposed mechanism consisted of oxidative addition of N-(ortho-iodo)aryl enamines to Pd(0), generating Pd(II) intermediate 335 which subsequently transformed into a seven-membered palladacycle intermediate 338 via intermediates 336 and 337. The latter underwent reductive elimination to give a new intermediate 339 which was converted to the desired product via [1,5]-H shift (Scheme 103).
 |
| | Scheme 102 | |
 |
| | Scheme 103 | |
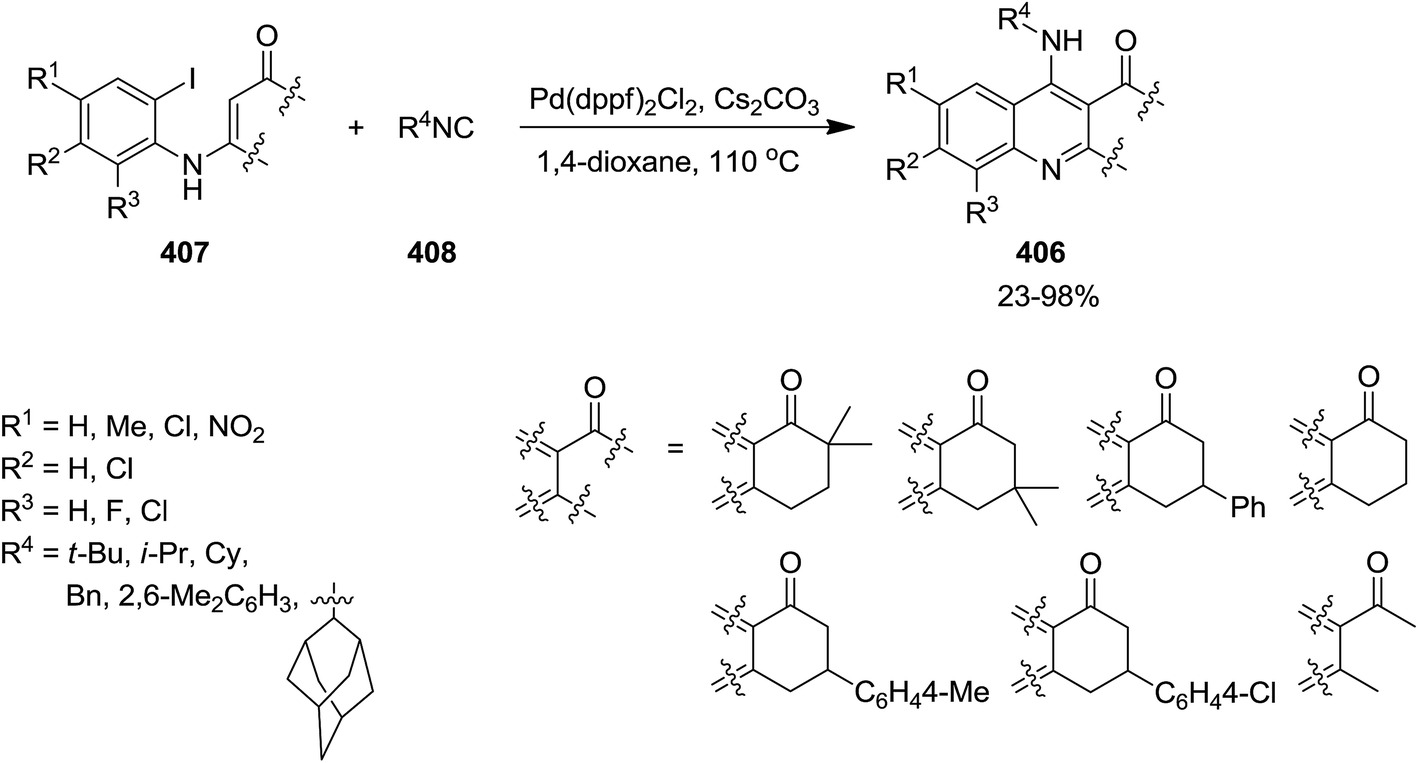
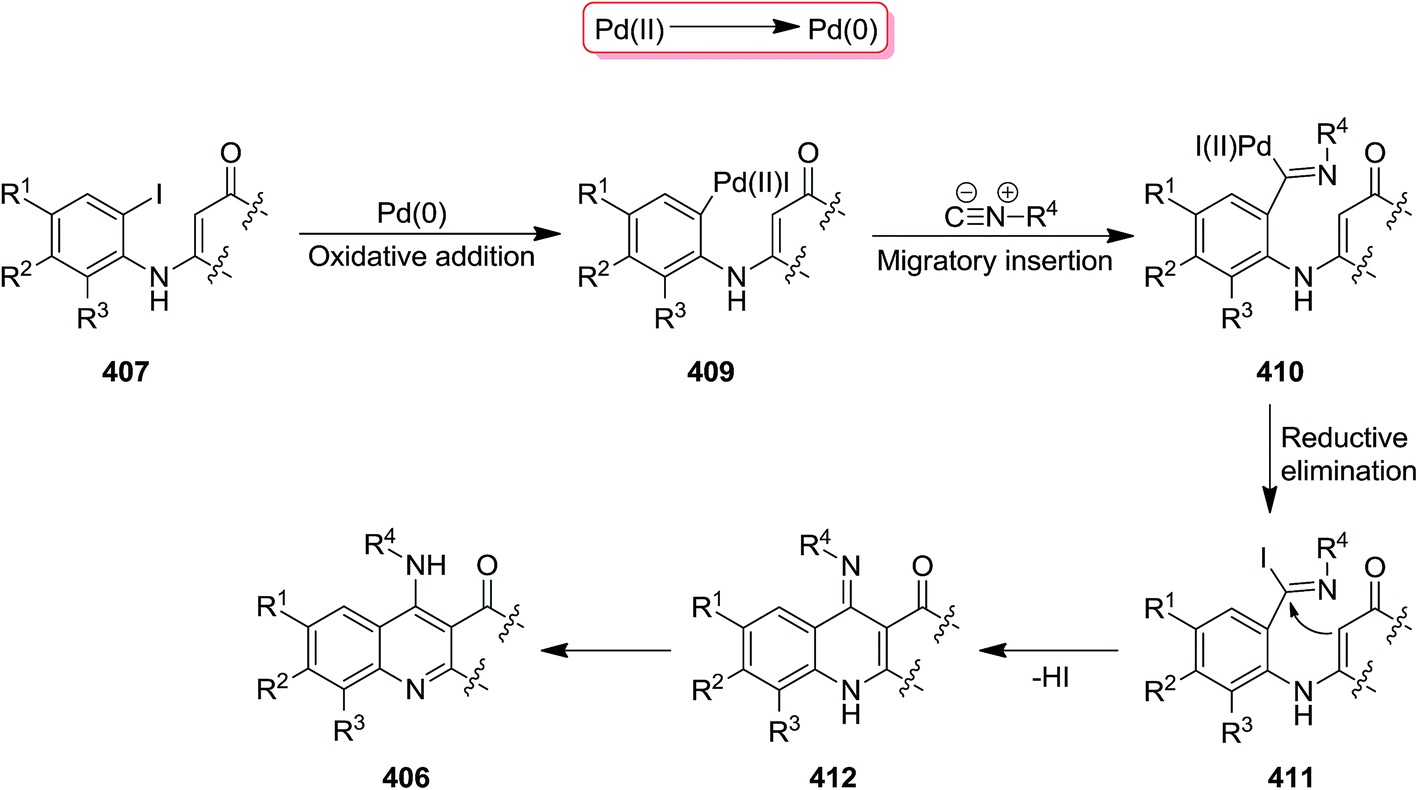
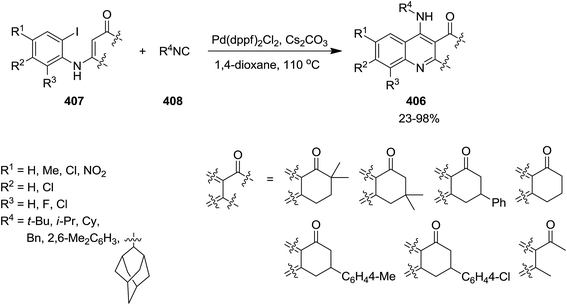
Pd was used as catalyst to promote the synthesis of 4-aminoquinolines 406 via a [5 + 1] annulation of tandem reaction between enaminones 407 and isocyanides 408 in 1.4-dioxane as solvent and in the presence of Cs2CO3 as a base (Scheme 104).225 Pd(dppf)2Cl2 showed superiority in terms of activity when compared with other Pd type catalysts including Pd(PPh3)2, Pd(dba)2, PdCl2, Pd(PPh3)2Cl2, and Pd(dppf)2Cl2. The proposed mechanism involves, initial oxidative addition of enaminones to the Pd(0) catalyst to form 409. A palladium(II) complex 410 is generated via isocyanide insertion, followed by reductive elimination to generate 411. The base played a crucial role in abstraction from C–H bond at α position of CO resulted in the formation of a six-membered ring complex. The desired product is finally obtained from tandem 1,5-H shift of intermediate 412 (Scheme 105).
 |
| | Scheme 104 | |
 |
| | Scheme 105 | |
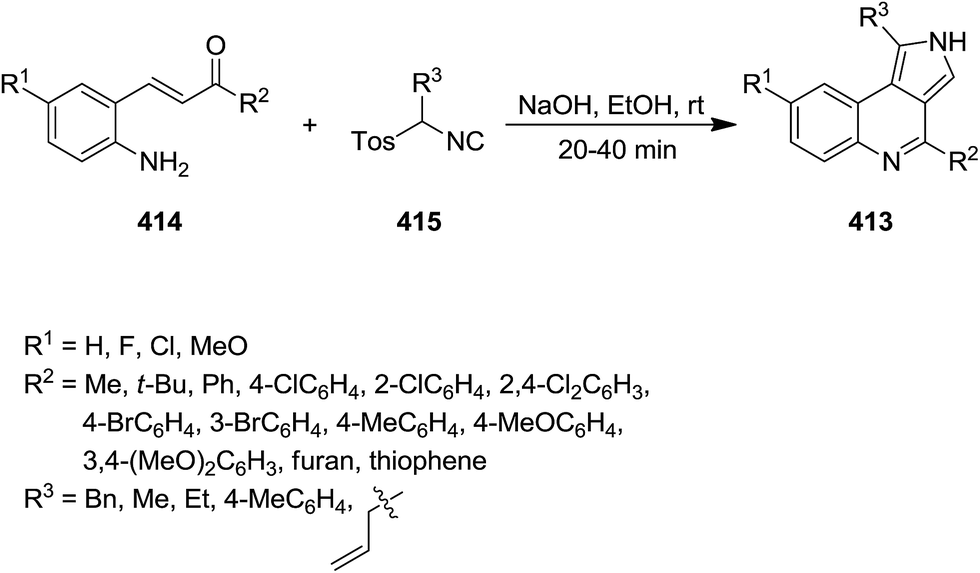
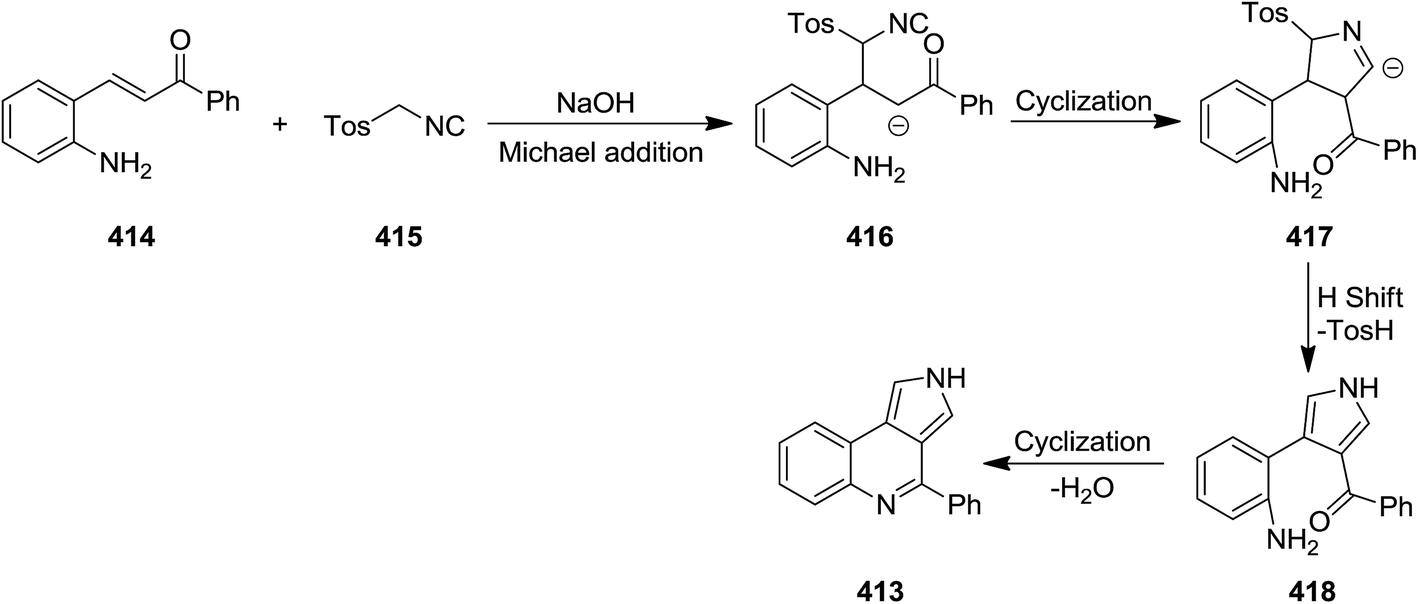
Tricyclic pyrrolo[3,4-c]quinoline derivatives 413 were synthesized efficiently via a rapid cascade reaction including [3 + 2] cycloaddition/cyclization of aminochalcones 414 and tosylmethyl isocyanides 415 under mild reaction condition (Scheme 106). To find the base of choice for this process, various bases such as Cs2CO3, NaOH, DBU, K2CO3, KOH were tested, among them the cost-effective and readily available NaOH proved to be the base of choice from different point of views. The process showed a good functionality tolerance and substrates. Both electron-donating and electron-withdrawing groups used, gave the desired products in high yields.226 The mechanism involved formation of carbanion 416 and its cyclization to form intermediate 417 followed by H-shift and cyclization of 418 (Scheme 107).
 |
| | Scheme 106 | |
 |
| | Scheme 107 | |
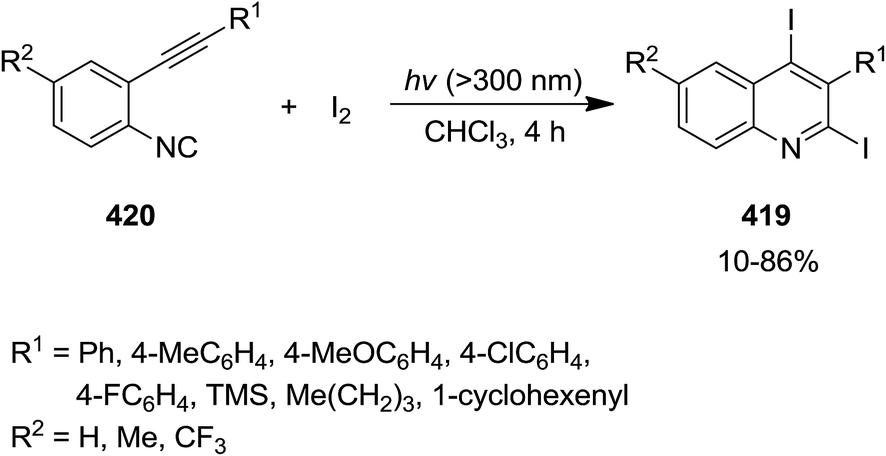
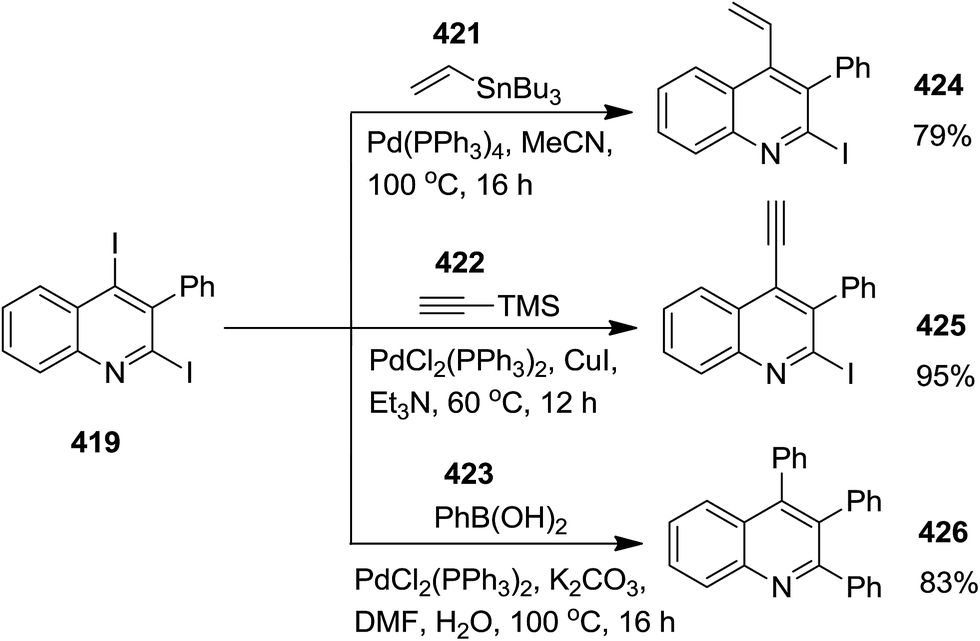
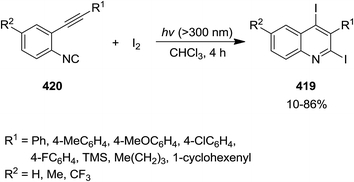
Iodine-promoted photochemical intramolecular annulation of o-alkynylaryl isocyanide derivatives 419 resulted in the formation of 2,4-diiodoquinolines 420 in high yield (Scheme 108). The synthetic broad scope of this process was established by using a wide variety of o-alkynylaryl isocyanide. The authors expanded their study, using the obtained products for various cross-coupling Pd-catalyzed name reactions (reaction with compounds 421–423), including Migita–Kosugi–Stille, Sonogashira and Suzuki–Miyaura to obtain poly-functionalized quinolines 424–426 (Scheme 109).227
 |
| | Scheme 108 | |
 |
| | Scheme 109 | |
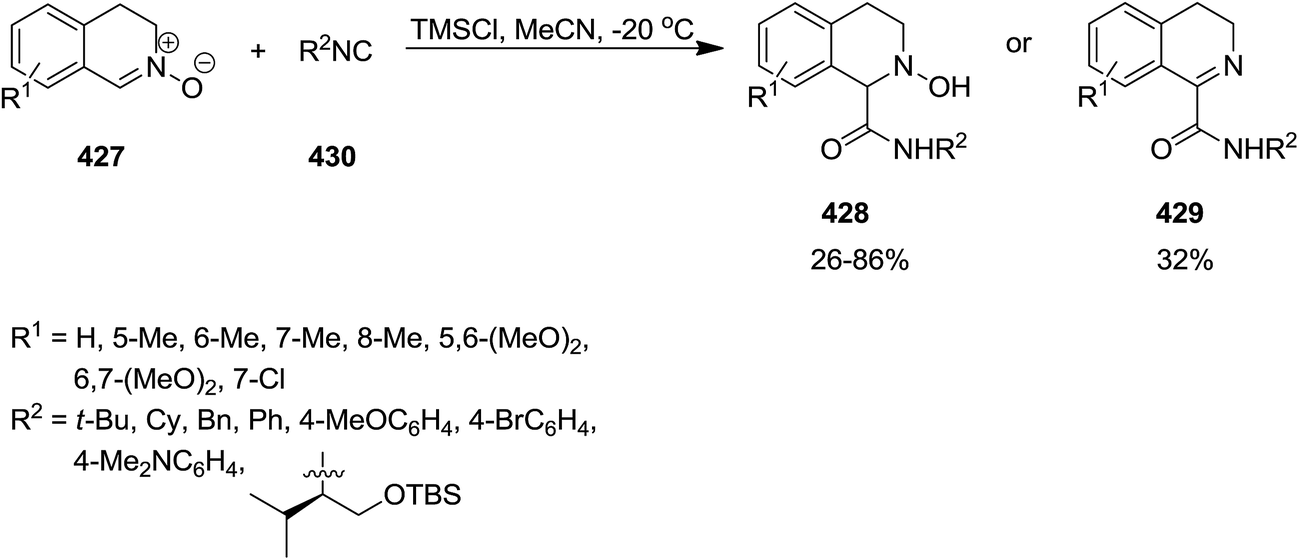
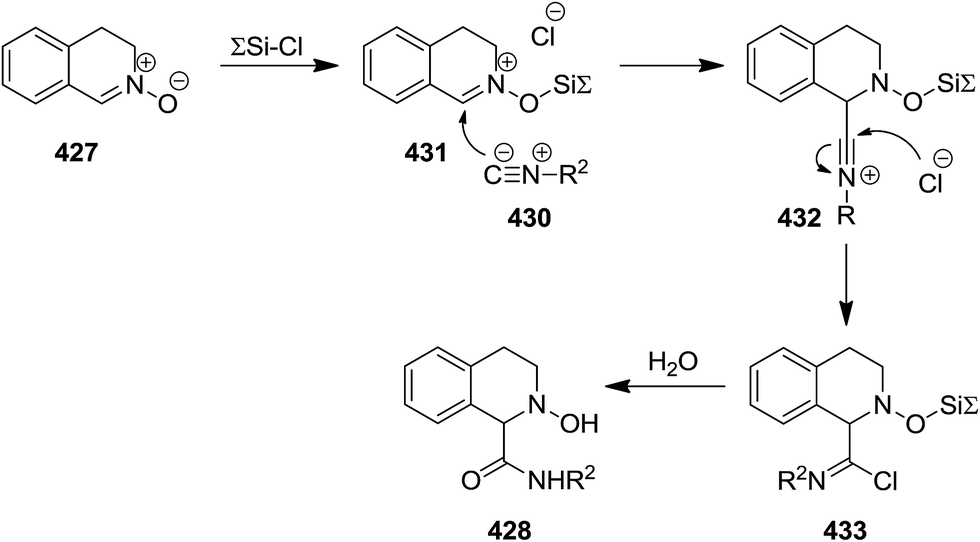
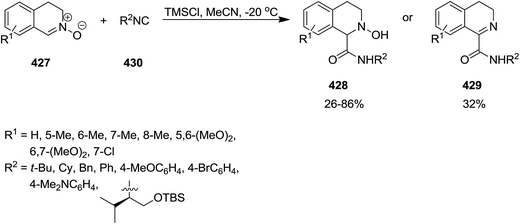
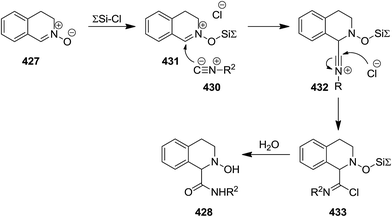
Chlorotrimethylsilane (TMSCl)-promoted isocyanide insertion to 3,4-dihydroisoquinoline N-oxide derivatives 427 resulted in the formation of 1,2,3,4-tetrahydroisoquinoline-1-carboxylamide 428 along with dehydrated compound 429 (Scheme 110). The scope of this process was studied by using various isocyanides 430 and nitrones. It was also disclosed that the presence of TMSCl is essential for the reaction to proceed smoothly. Furthermore, the effect of the nature of chlorosilane was investigated by using several chlorosilane derivatives such as sterically hindered ones i.e., tert-butylchlorodimethylsilane. It was found that the lesser steric hindrance at Si atom, resulted in the higher yields of products. The plausible mechanism, illustrated in Scheme 109, was based on coordination of O atom of nitrone with TMSCl leading to its activation and formation of 431. Furthermore, the generated nitrilium intermediate 432 can be stabilized via Cl− addition, followed by formation of imidoyl chloride 433. Hydrolyzation of the latter furnished the final product (Scheme 111).228
 |
| | Scheme 110 | |
 |
| | Scheme 111 | |
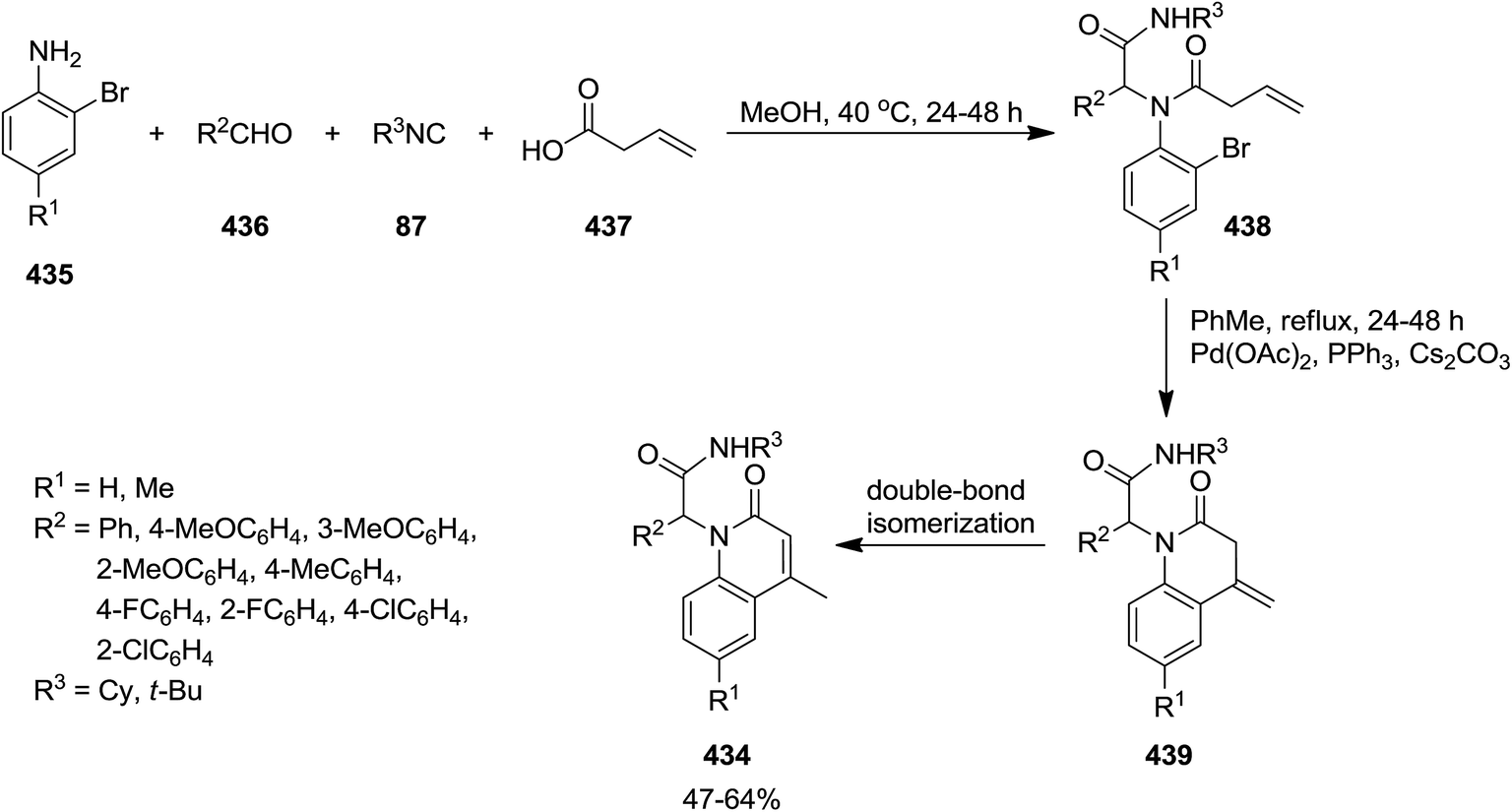
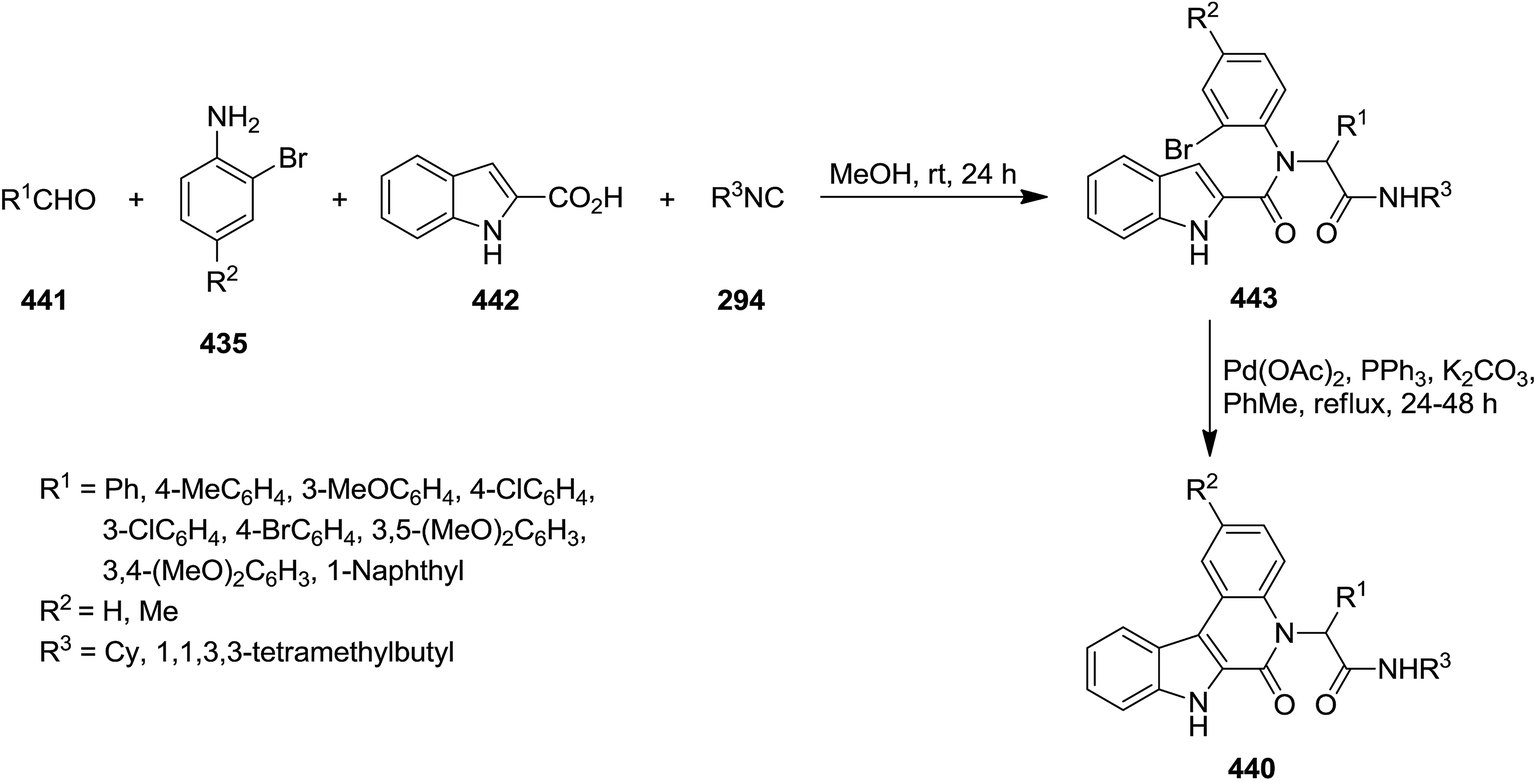
The two-step synthesis of new N-substituted-4-methyl-quinolin-1(2H)-ones 434 was reported by Foroumadi et al. initially, four-component Ugi reaction of 2-bromoanilines 435, aromatic aldehydes 436, vinylacetic acid 437, and isocyanides 87 resulted in acyclic Ugi adducts 438 which was subjected to Pd-catalyzed intramolecular Heck reaction to afford the desired corresponding products 439 in the presence of Pd(OAc)2 in high yields.229 The generality of this procedure was confirmed by using differently substituted aromatic aldehydes bearing electron donating and electron withdrawing functional groups (Scheme 112).
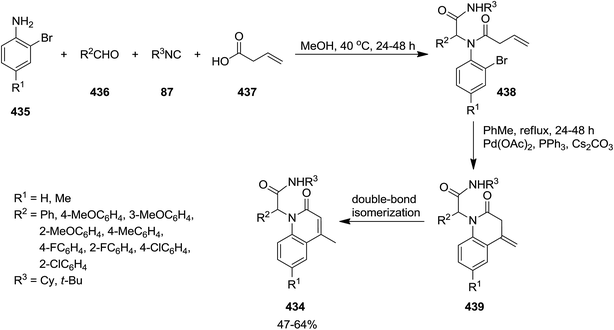 |
| | Scheme 112 | |
Efficient synthesis of indolo[2,3-c]quinolinones 440 was reported via four-component Pd-catalyzed Ugi reaction of 2-bromoanilines 435, aromatic aldehydes 441, isocyanides 294 and indole-2-carboxylic acid 442 through intermediate 443 (Scheme 113).230 The broad substrate scope, simplicity and excellent yields were claimed as the advantages of this synthetic strategy. Studying the reaction variables such as solvent and catalyst type provided the optimized reaction conditions.
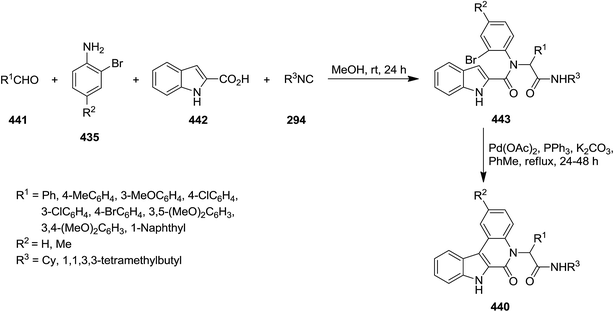 |
| | Scheme 113 | |
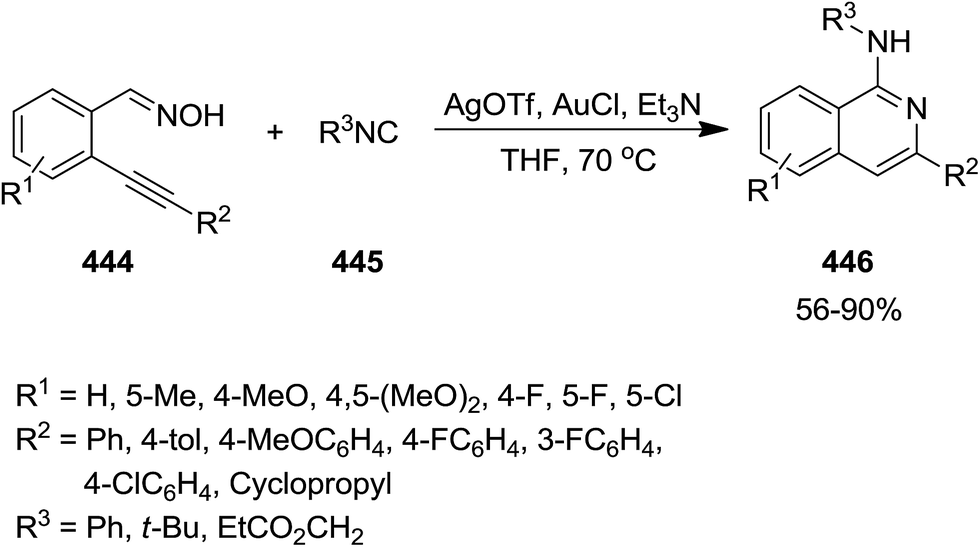
Silver triflate and gold(I)chloride were exploited as catalyst to promote the cascade reaction of 2-alkynylbenzaldoximes 444 and 2-isocyanoacetates 445 to obtain 1-aminoisoquinoline derivatives 446 (Scheme 114).231 This work could provide insight into transition metal-catalyzed isocyanide activation chemistry. It was demonstrated that the using weak base and using another metal as co-catalyst promoted the yield of reaction, remarkably. Combining silver triflate with various Lewis acids was not promising while using gold(I)chloride which could generate gold(I)triflate in the presence of silver triflate led to the desired products in good to high yields. Noticeably the use of gold(I)triflate as sole catalyst was less efficient compared with using two-metal catalyst and co-catalyst system.
 |
| | Scheme 114 | |
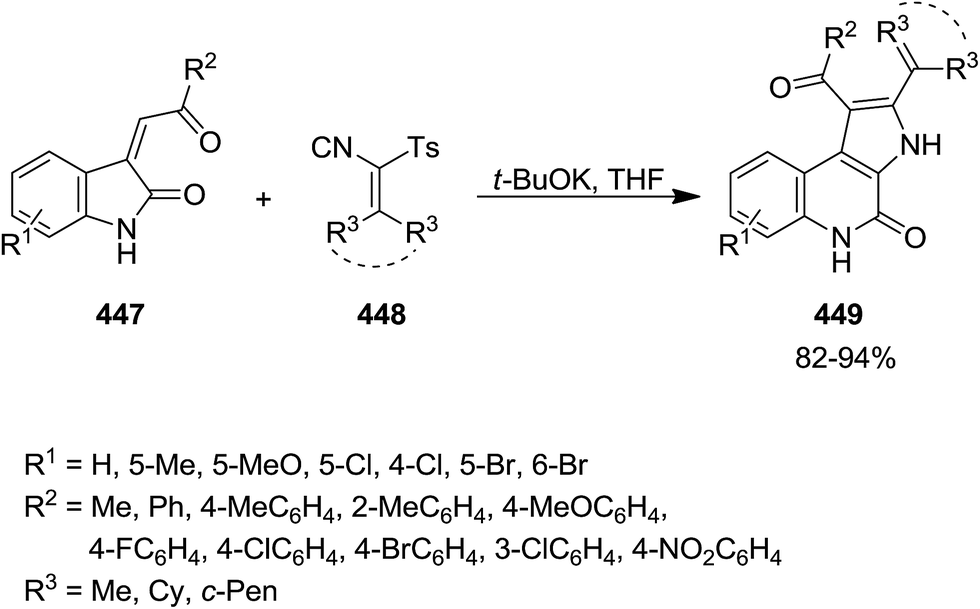
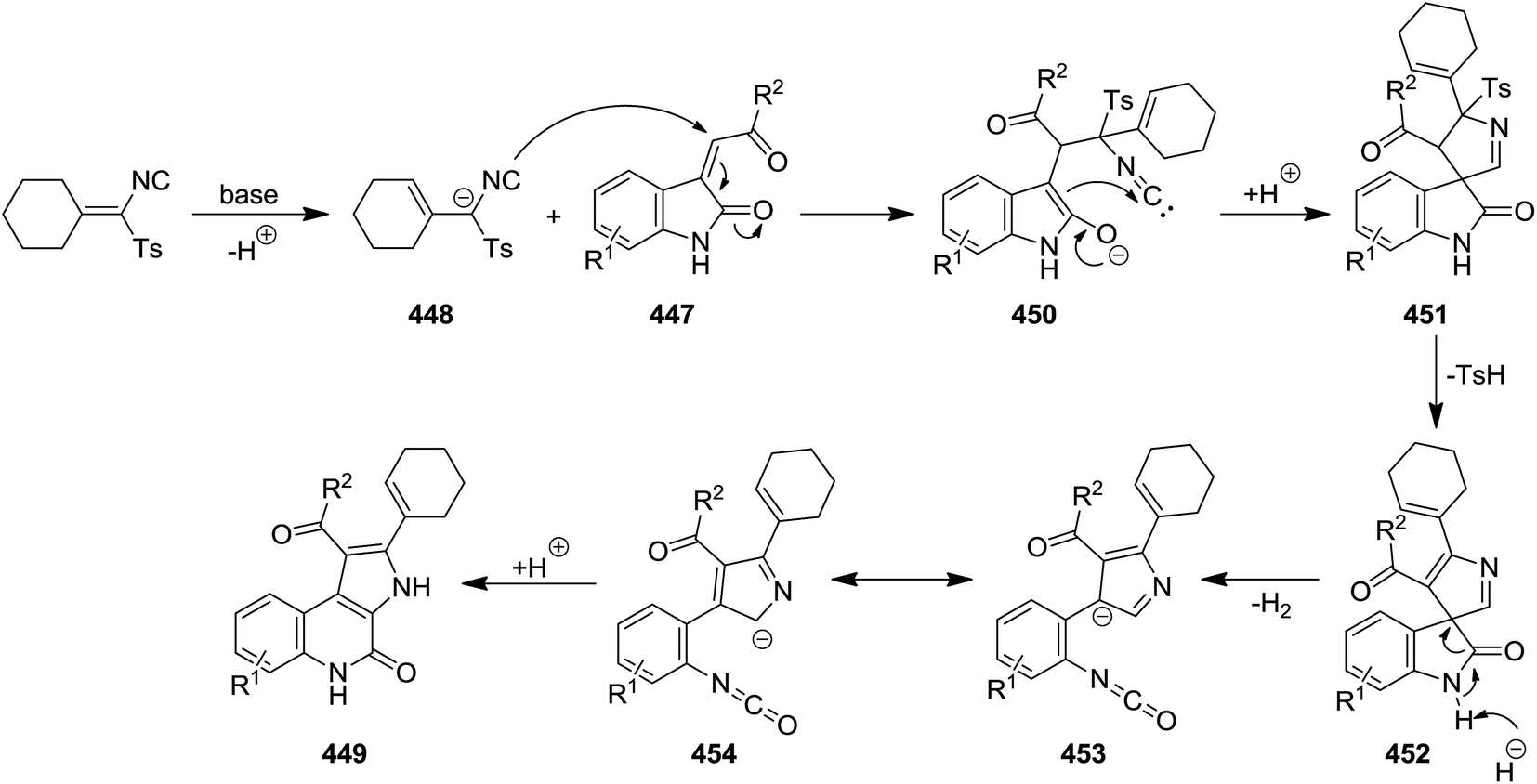
The reaction of (Z)-3-(2-oxo-2-ethylidene)indolin-2-ones 447 and TosMIC 448 in THF and in the presence of t-BuOK as base proceed smoothly to afford the corresponding 3H-pyrrolo[2,3-c]quinolin-4(5H)-one tosylmethyl 449 (Scheme 115).232 The synthetic scope of this reaction was confirmed by using (Z)-3-(2-oxo-2-ethylidene)indolin-2-one derivatives 447 with various electron-donating and electron withdrawing groups as well as cyclic and acyclic TosMICs. The proposed mechanism involves the deprotonation of TosMIC generating an anion 450 at α-position of isocyanide, followed by migration of CC double bond. The CC double bond in (Z)-3-(2-oxo-2-ethylidene)indolin-2-one can be subjected to nucleophilic addition, generating an intermediate 451. The intramolecular reaction of isocyanide moiety led to ring closure and formation of a new intermediate which by removal of tosyl group afforded a spiro intermediate 452. The latter underwent ring opening and loss of proton to produce 453, followed by tautomerization. The desired product was achieved via nucleophilic attack of carbon anion of 454 to isocyano group with simultaneous ring closing (Scheme 116).
 |
| | Scheme 115 | |
 |
| | Scheme 116 | |
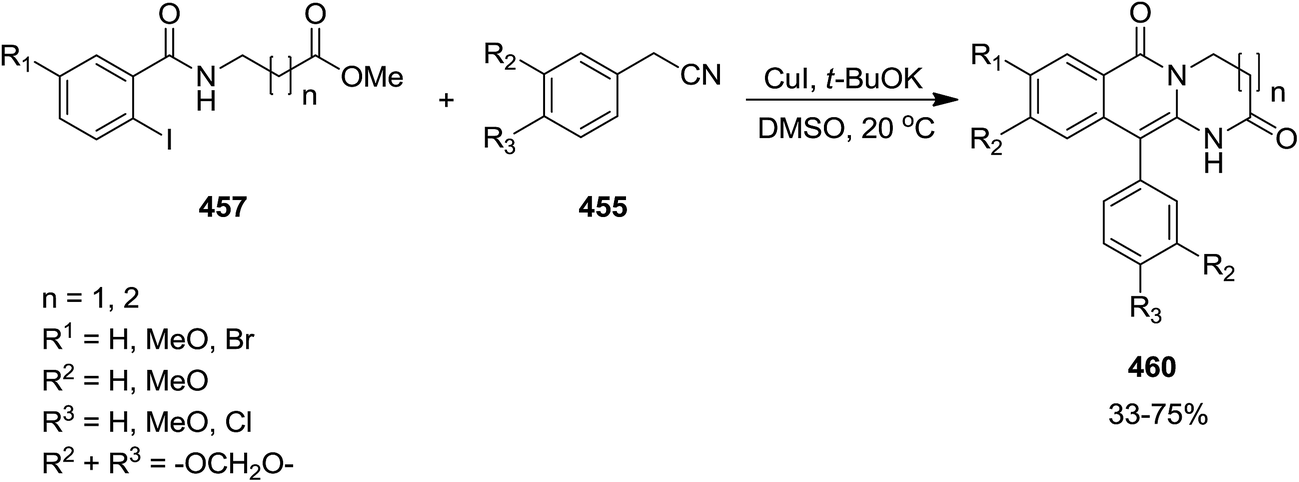
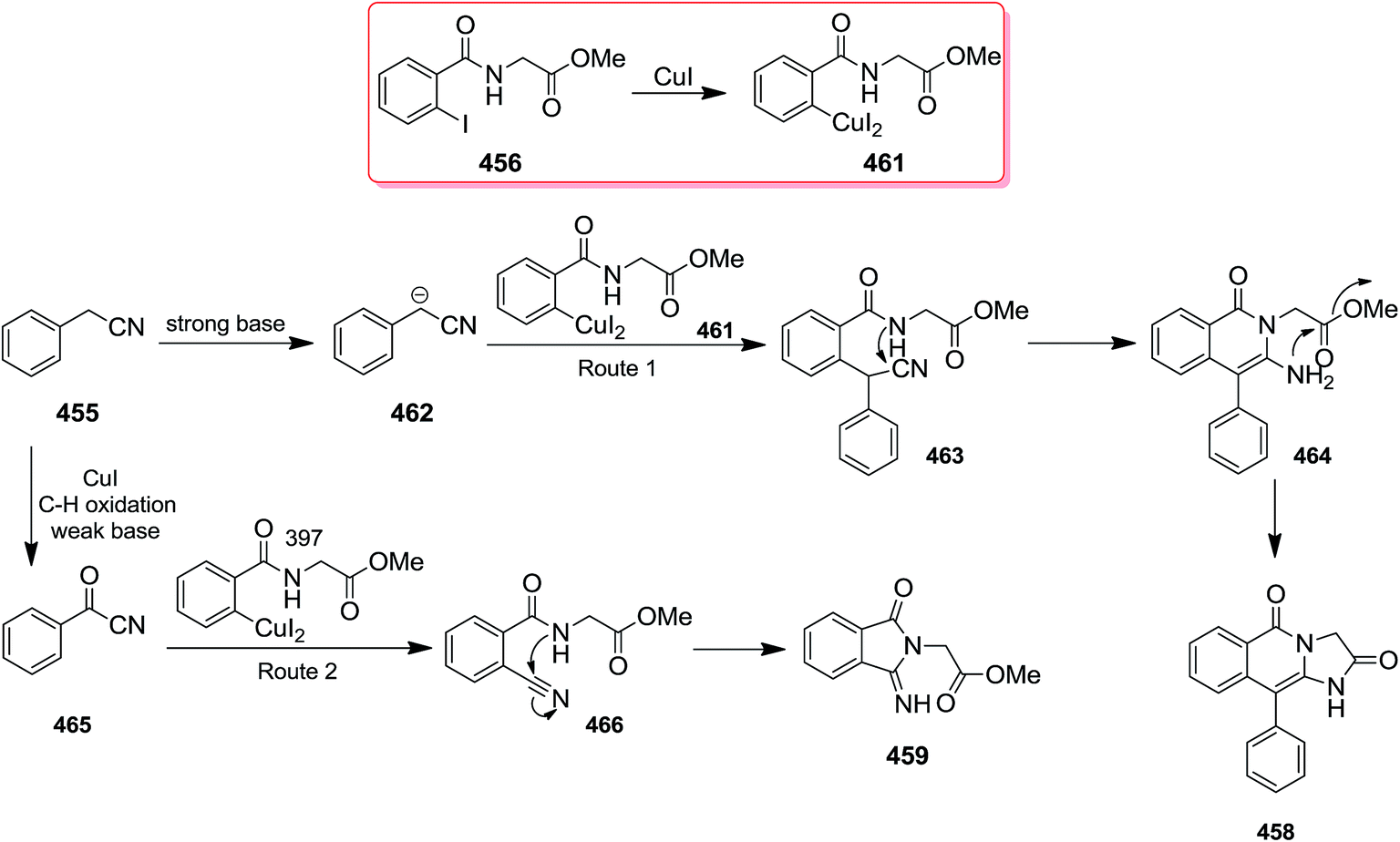
2.3.3 Isoquinolinones. The copper(I)iodide catalyzed reaction of benzyl cyanide 455 with 2-halobenzamides of amino acid esters 456 and 457 resulted in the corresponding isoquinoline 458, 459 and 460. The reaction conditions in this approach is vital since strong basic conditions resulted in fused isoquinolinones, while using weak base led to the formation of iminoisoindolinone (Schemes 117 and 118).233 The reaction mechanism is depicted in Scheme 119. The mechanism involved formation of species 458. In the presence of strong base carbanion 462 would form and reacted with 461 to form 463 which subsequently formed 464. In the presence of weak base, however, oxidation would generate 465 which reacted with 461 to form 466. It was found that replacing copper(I)iodide with copper(I)chloride furnished the desired product in virtually similar yields whereas, the reaction failed when copper(I)oxide was employed. Good to high yields, broad substrate scope and mild reaction conditions were the merits, claimed for this strategy.
 |
| | Scheme 117 | |
 |
| | Scheme 118 | |
 |
| | Scheme 119 | |
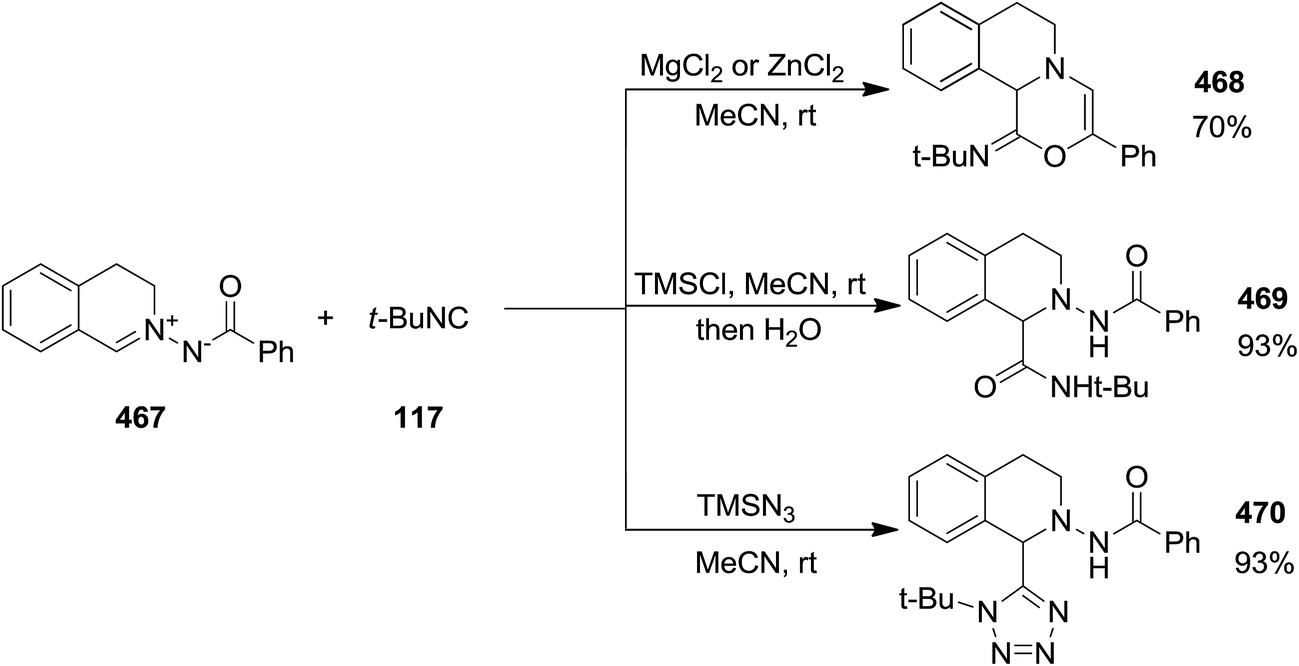
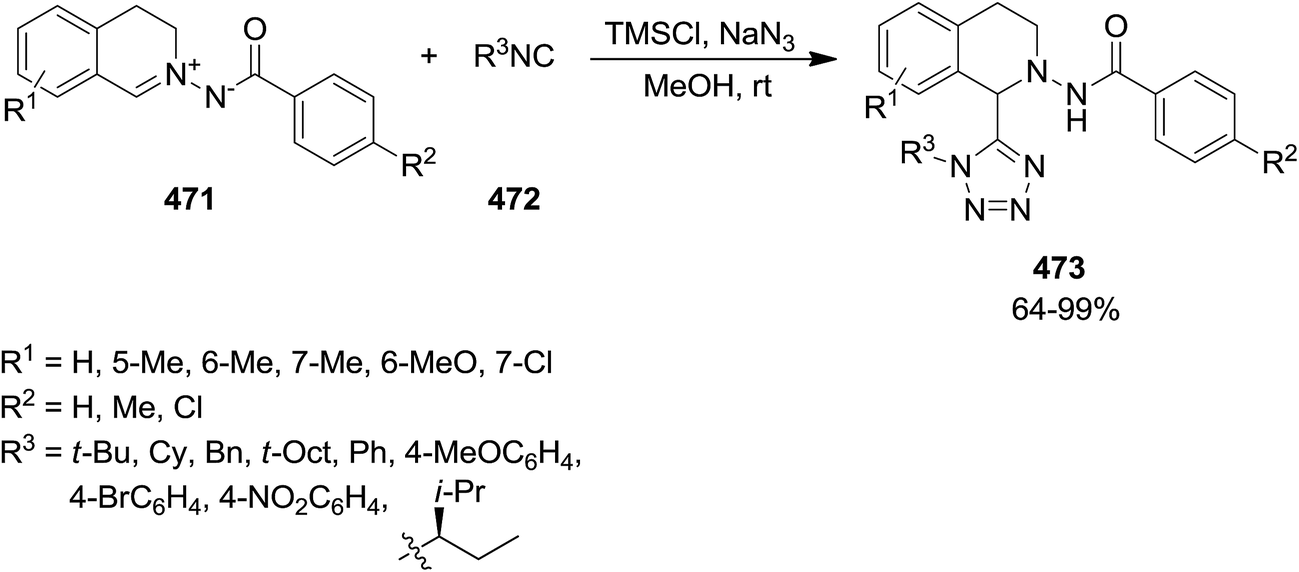
The reaction of C,N-cyclic N′-acyl azomethine imines 467 or 471 and isocyanides 117 and 472 in the presence of MgCl2 or ZnCl2 led to the formation of imin-1,3,4-oxadiazin-6-ones 468 while using chlorotrimethylsilane, TMSCl, by preventing the cyclization, resulted in 1,2,3,4-tetrahydroisoquinoline-1-carboxylamide derivatives 469 in high yields. 1,2,3,4-Tetrahydroisoquinoline-1-carboxylamides 470 and 473 were the main product of this reaction in the presence of trimethylsilyl azide, TMSN3 (Scheme 120). Remarkably, the mixture of TMSCl and sodium azide as a less expensive alternative of TMSN3 can also be successfully used (Scheme 121).234 The presence of electron-withdrawing functionalities on aromatic moiety of C,N-cyclic N′-acyl azomethine imines resulted in the requirement of longer reaction times as well as obtaining lower yields. Furthermore, benzoyl moiety bearing Me substituent was found a better substrate than the corresponding Cl substituent in terms of reaction yields and reaction times.
 |
| | Scheme 120 | |
 |
| | Scheme 121 | |
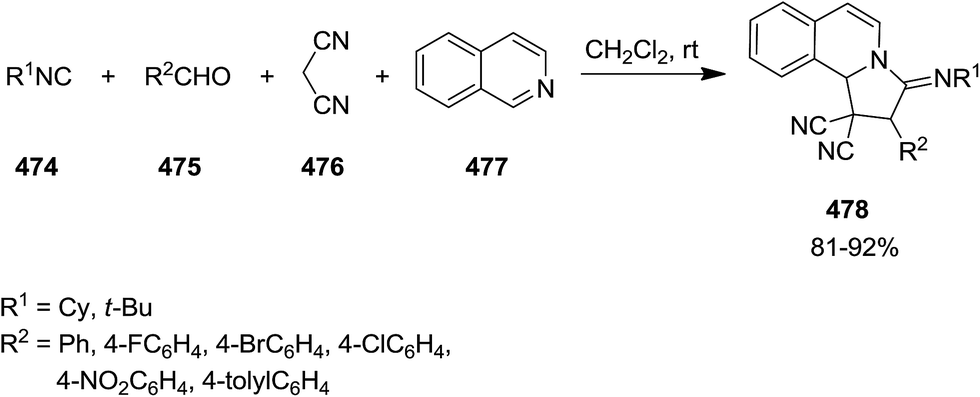
A series of 3-(t-butylimino or cyclohexylimino)-2-aryl-2,3-dihydropyrrolo[2,1-a]isoquinoline-1,1(10b H)-dicarbonitrile 478 were obtained in high yields from multi-component reaction of isocyanides 474, aldehydes 475, malononitrile 476, and isoquinoline 477 (Scheme 122).235 The proposed reaction mechanism was based on formation of a Knoevenagel intermediate 479 which furnished compound 480. Subsequently, compound 481, generated through addition reaction of 480 with isoquinoline 477 which furnished the desired product trough cyclization (Scheme 123). Mild reaction condition and broad substrate scope were the merits of this protocol.
 |
| | Scheme 122 | |
 |
| | Scheme 123 | |
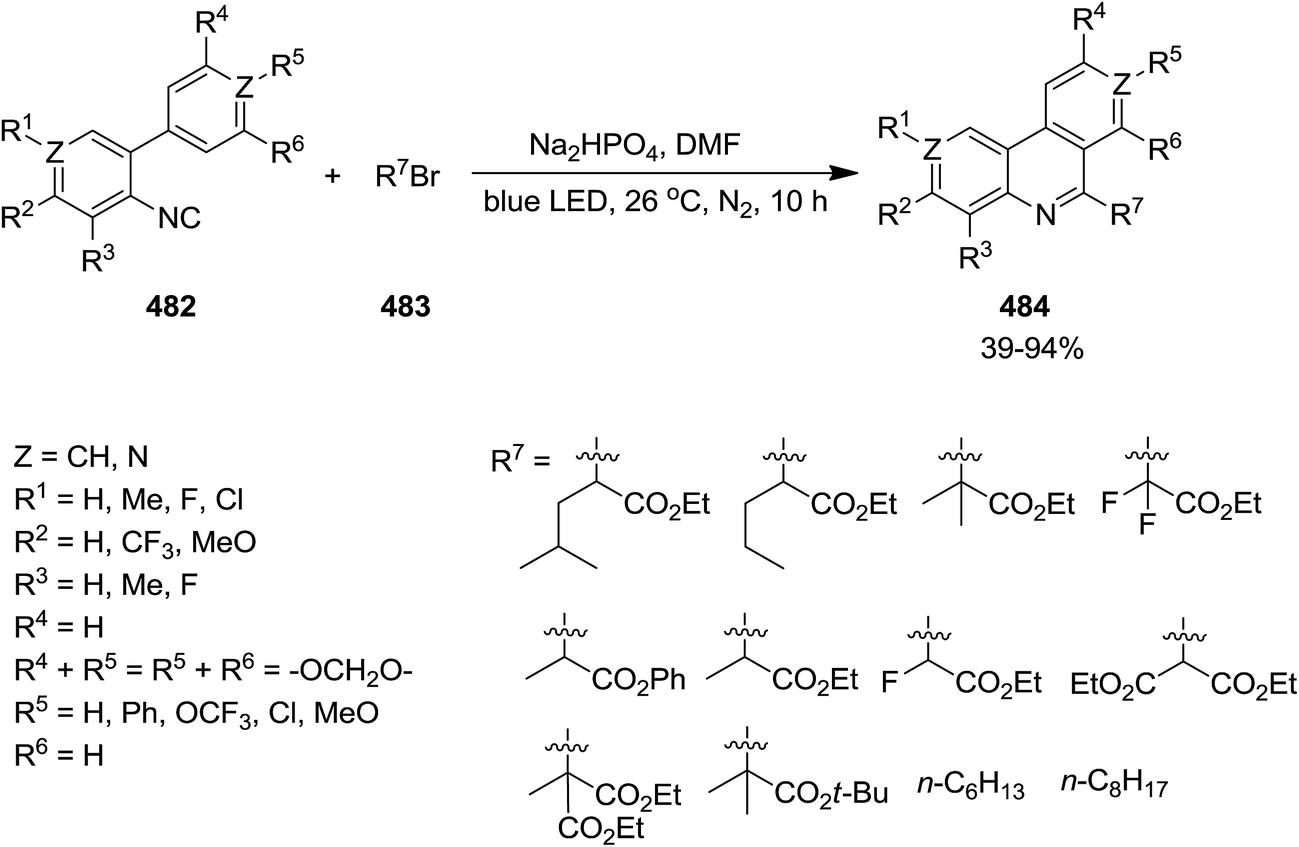
2.3.4 Phenanthridine. Photo-assisted somophilic isocyanide 482 insertion to 483 was utilized to provide 6-alkylatedphenanthridines 484 in good to high yields (Scheme 124).236 This synthetic method can be considered as an efficient, cost-effective, environmentally benign and mild protocol for the synthesis of broad range of phenanthridines. Moreover, this process requires no external oxidant and only gave HBr as by-product which could be easily handled.
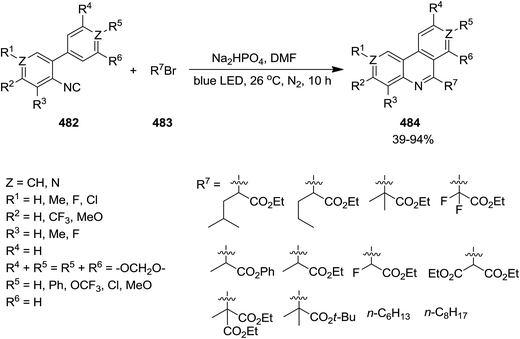 |
| | Scheme 124 | |
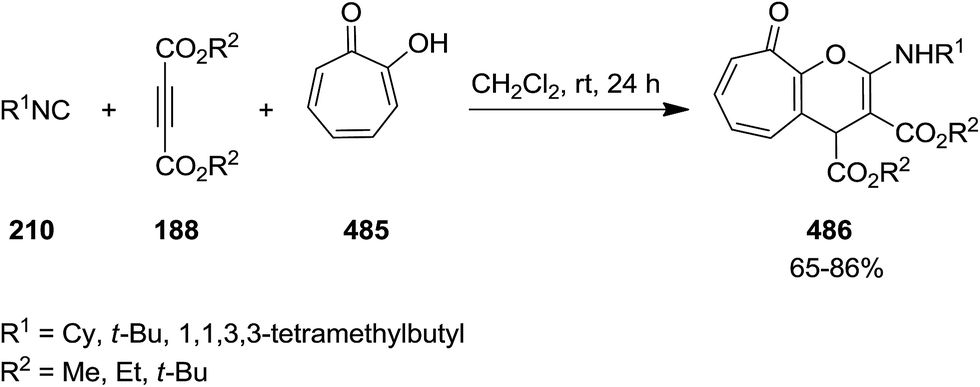
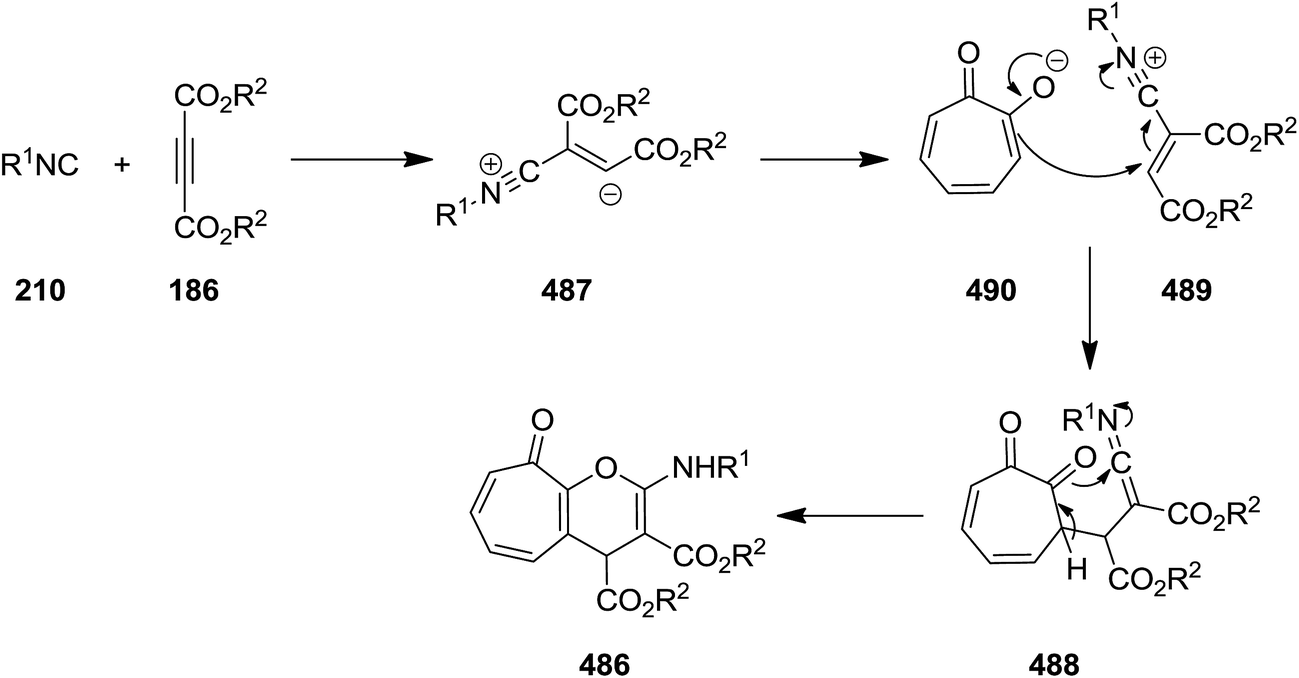
2.3.5 Pyran. Catalyst-free three-component reaction of α-tropolone 485, dialkyl acetylenedicarboxylate 188 and alkyl isocyanide 210 proceeded smoothly at room temperature to give dialkyl 2-(alkylamino)-4,9-dihydro-9-oxocyclohepta[b]pyran-3,4-dicarboxylate derivatives 486 in high yields (Scheme 125).237 The plausible mechanism involves the reaction of dialkyl acetylenedicarboxylate and alkyl isocyanide to give a zwitterionic intermediate 487 which is protonated. Subsequently, a ketenimine 488 is generated from the reaction of nitrilium ion 489 and deprotonated tropolone 490 which undergoes tautomerization and annulations to afford the final product (Scheme 126).
 |
| | Scheme 125 | |
 |
| | Scheme 126 | |
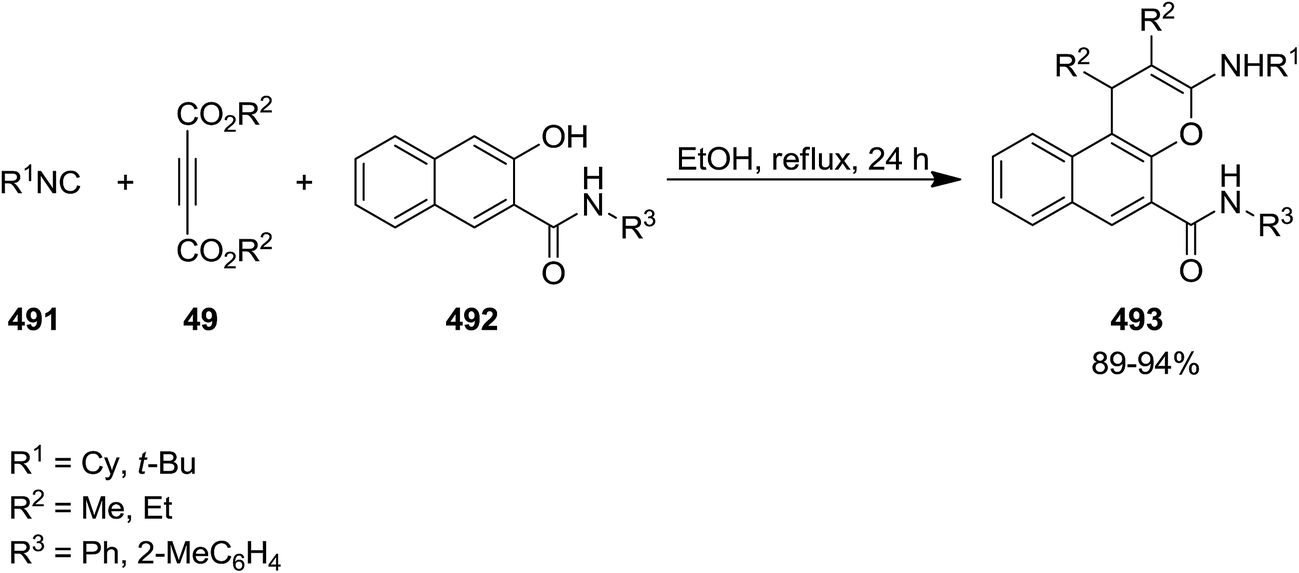
2.3.6 Chromene. Reaction of dialkyl acetylenedicarboxylates 49, alkyl isocyanides 491 and N-aryl-3-hydroxynaphthalene-2-carboxamide 492 resulted in the formation of substituted 4H-chromene derivatives 493 after 24 h. Broad scope of substrate, high yields, neutral condition and simplicity of work up were mentioned as the merits of this synthetic approach (Scheme 127).238 The reaction commenced with isocyanide insertion to dialkyl acetylenedicarboxylate, followed by protonation. The later reacted with conjugate anion of N-aryl-3-hydroxynaphthalene-2-carboxamide to afford a ketenimine which subjected to cyclization to furnish the final product.
 |
| | Scheme 127 | |
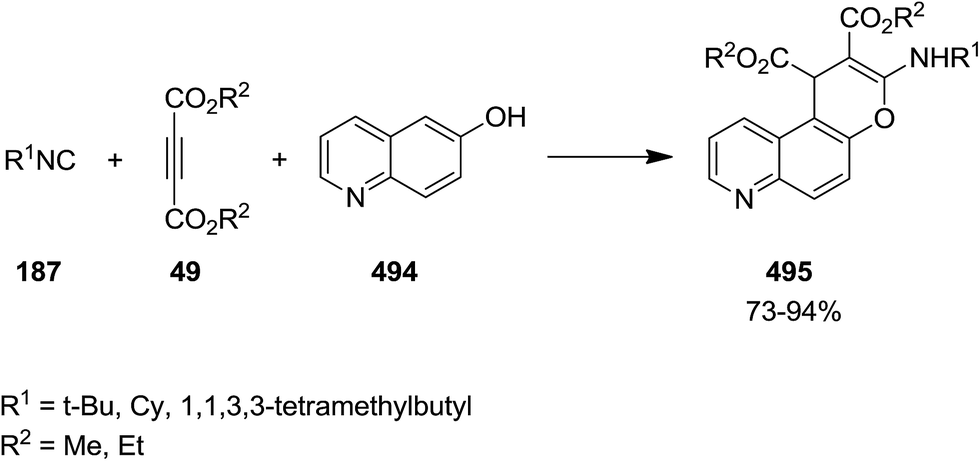
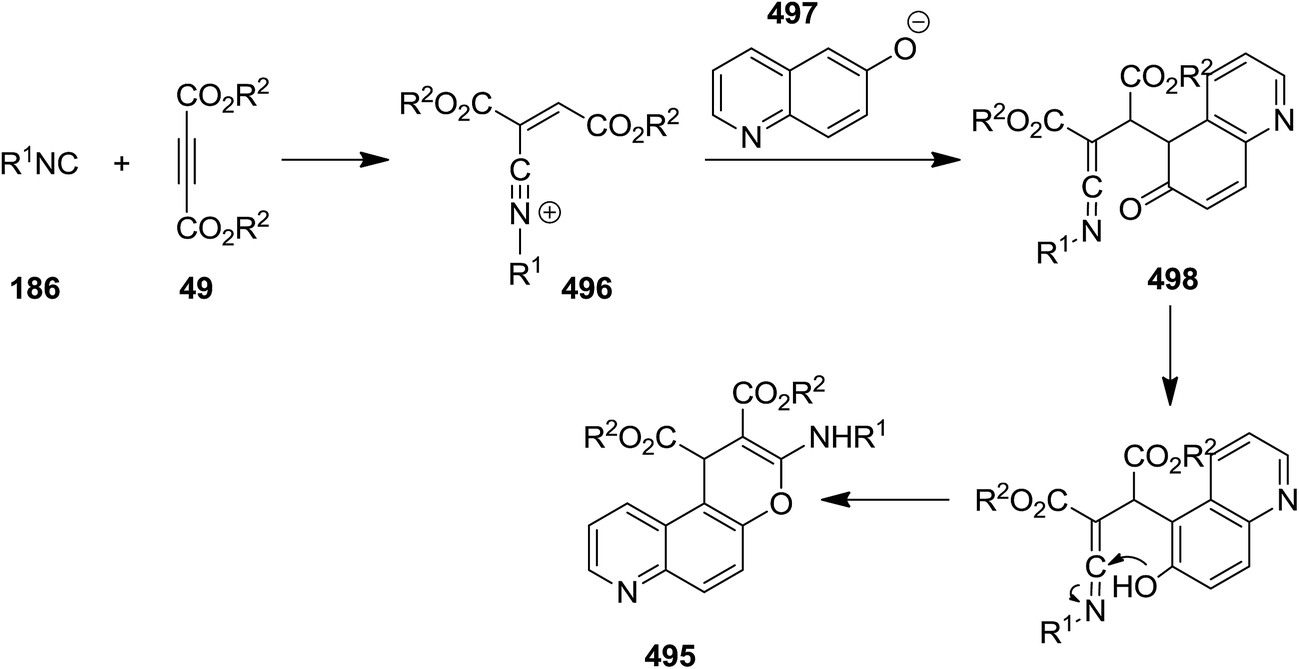
Poly-substituted 4H-chromene derivatives 495 were synthesized efficiently using three-component reaction of dialkyl acetylenedicarboxylate 49, alkyl isocyanides 187 and 6-quinolinol 494 (Scheme 128). The plausible mechanism involves the formation of an intermediated via isocyanide insertion to dialkyl acetylenedicarboxylate, followed by protonation of the obtained adduct 496. A ketenimine 498 then is generated from reaction with anion 497. Finally, tautomerization and cyclization afford the final product (Scheme 129).239
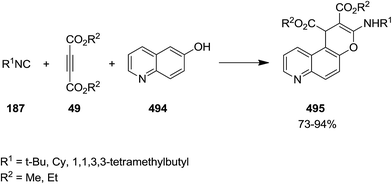 |
| | Scheme 128 | |
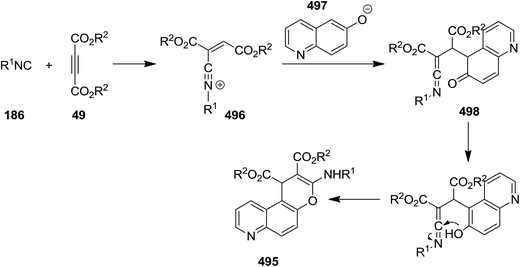 |
| | Scheme 129 | |
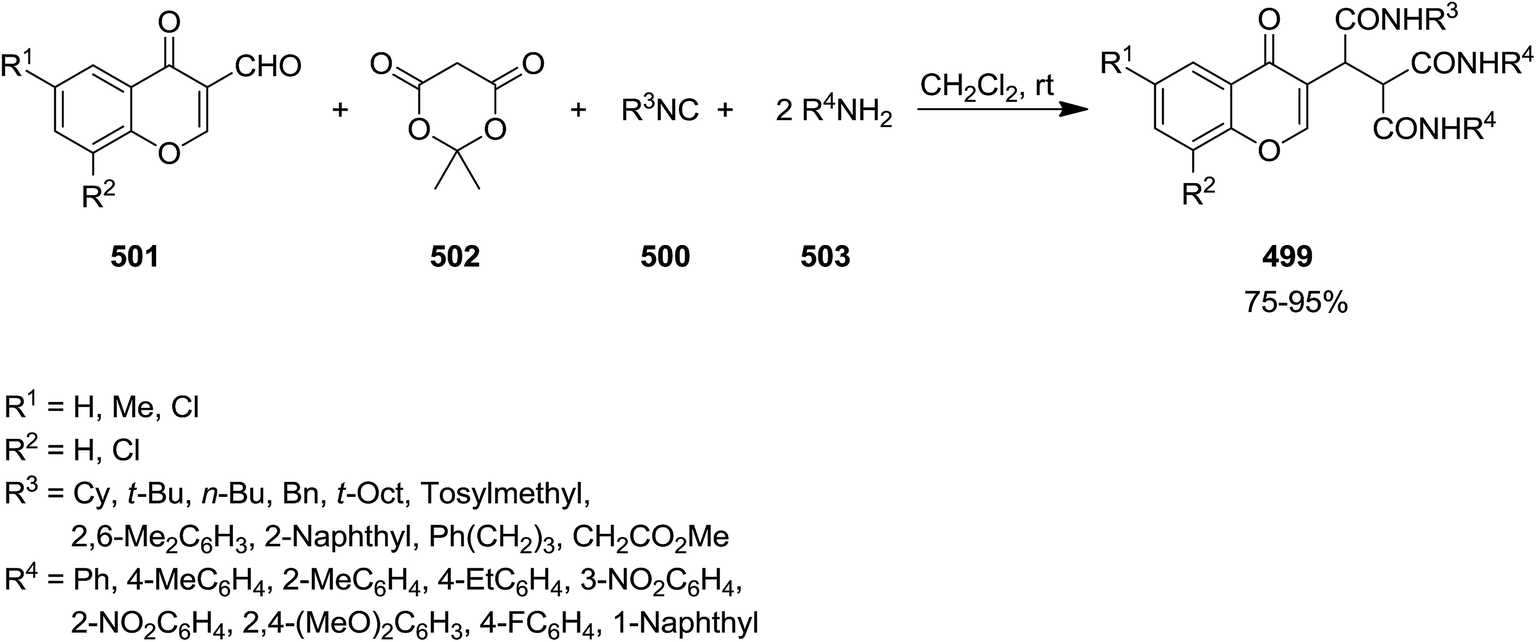
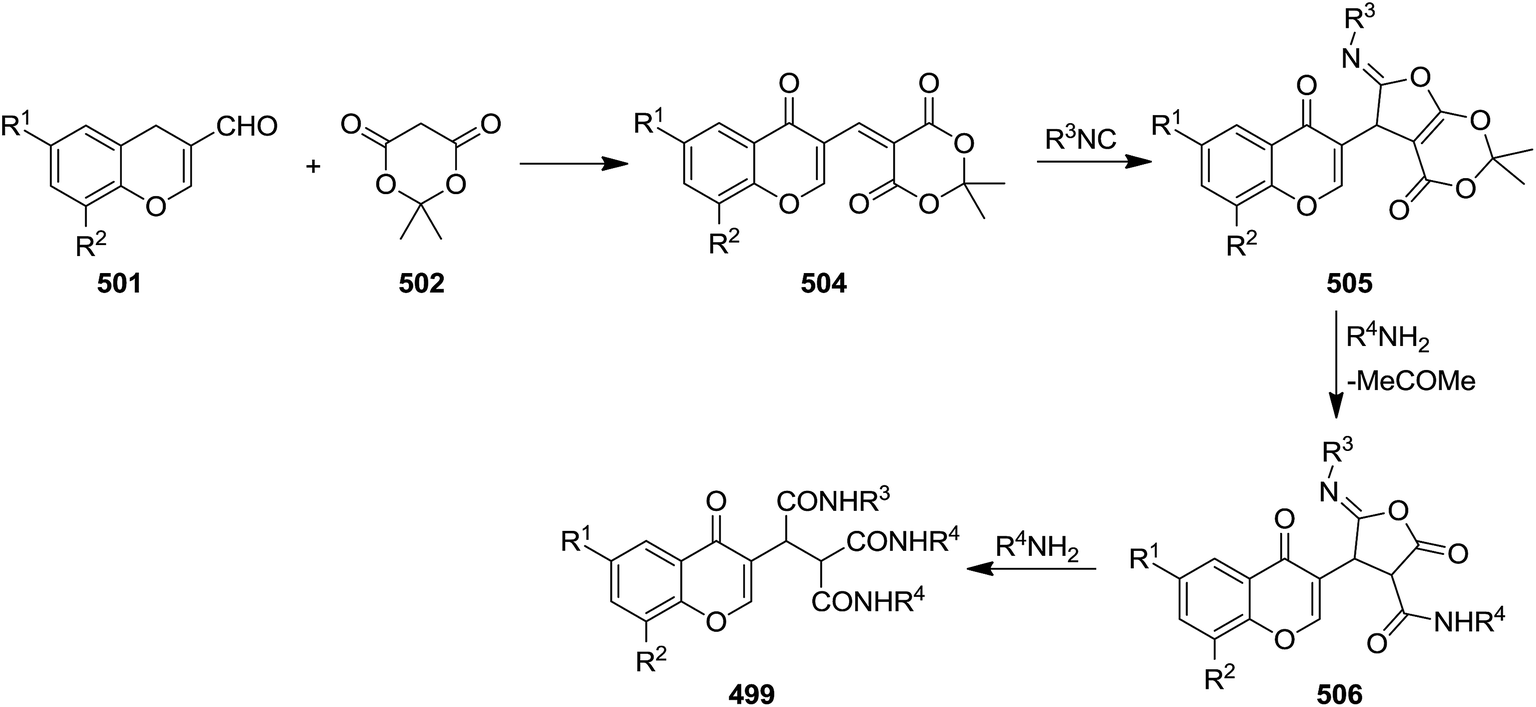
A novel isocyanide-based synthetic strategy was developed for the preparation of chromones derivatives containing tripeptides 499. The process involved pseudo-five-component condensation of isocyanides 500, 3-formylchromones 501, Meldrum's acid 502 and primary aromatic amines 503 in CH2Cl2 at ambient temperature (Scheme 130).240 The formation of all new bonds, i.e. two C–C, CO and two C–N occurred concomitantly. The reaction could proceed via Knoevenagel condensation of Meldrum's acid and 3-formylchromones and formation of 504. The obtained adduct 504 underwent [1 + 4] cycloaddition reaction with isocyanide to furnish an intermediate 505. The reaction of the latter with arylamine followed by removal of acetone resulted in production of an iminolactone 506 which would be subjected to nucleophilic attack of another arylamine to afford final product (Scheme 131).
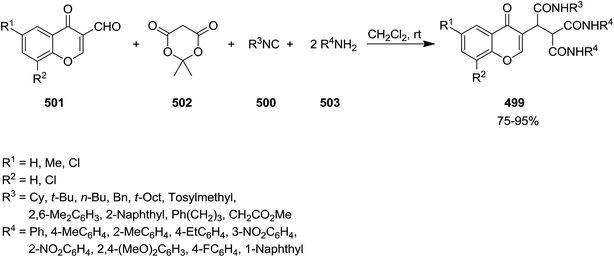 |
| | Scheme 130 | |
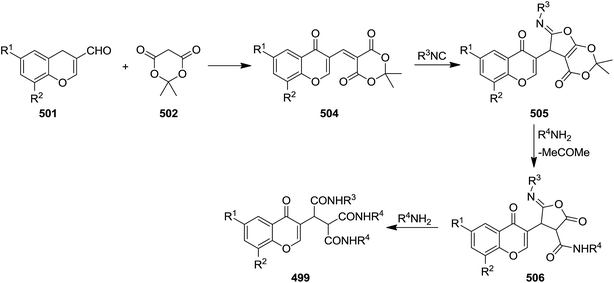 |
| | Scheme 131 | |
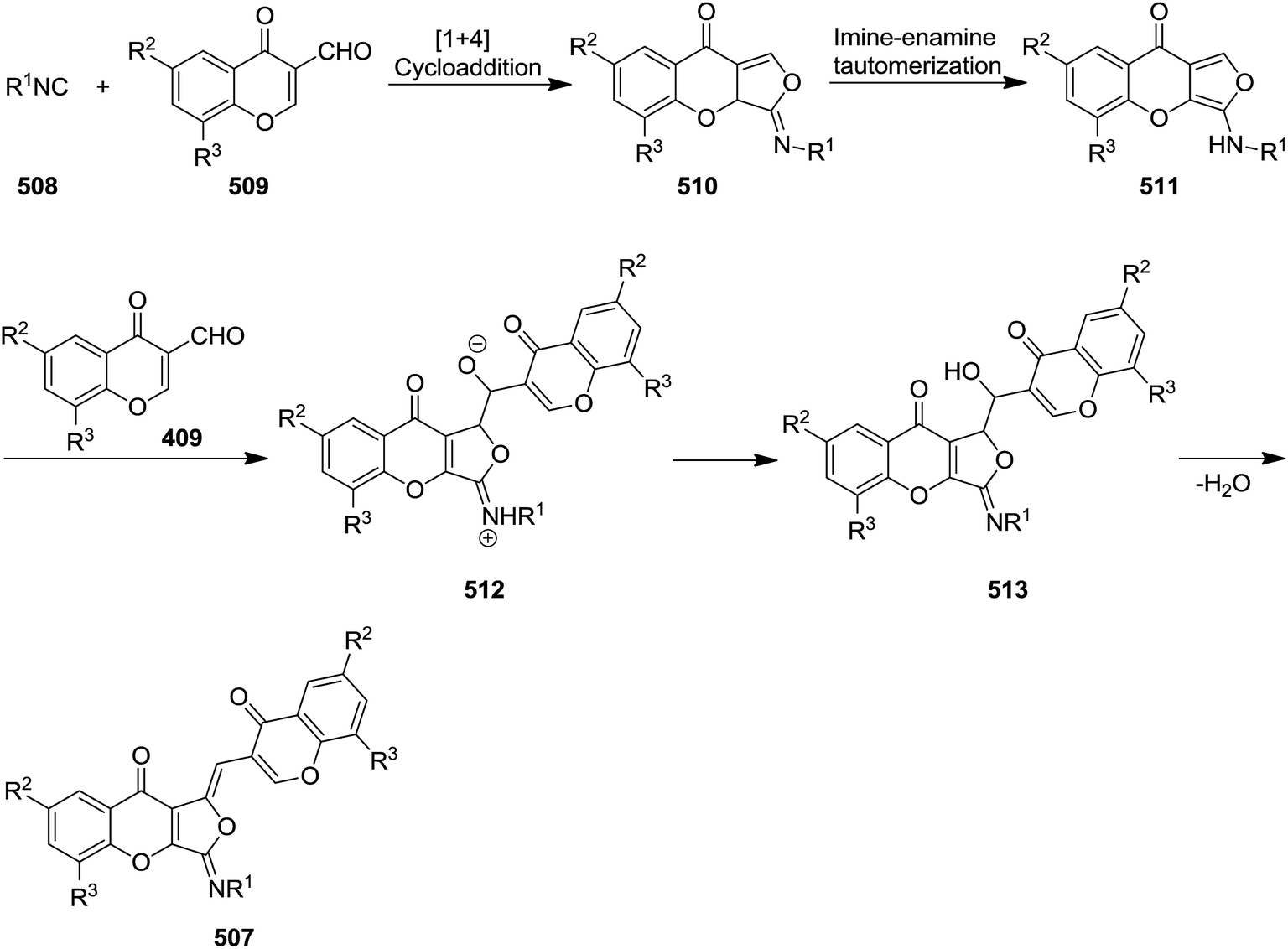
A series of organic fluorophores (1Z)-3-(alkylimino)-1-[(chromone-3-yl)methylene]-1,3-dihydro-9H-furo[3,4-b]chromen-9-one derivatives 507 were synthesized via one-pot reaction of alkyl isocyanides 508 and 2 equivalent 3-formylchromones 509 in anhydrous dichloromethane under mild reaction conditions (Scheme 132).241 It is worth noting that replacing alkyl isocyanides with aryl counterparts or replacing 1 equivalent of 3-formylchromones with other aldehyde did not lead to the desired products. The reaction proceeded via formation of a highly reactive fused 3-(alkylamino)-9H-furo[3,4-b]chromen-9-one intermediate 510 via the [4 + 1] cycloaddition of alkyl isocyanide to 3-formylchromone followed by imine–enamine tautomerization and formation of 511. The latter attacked the formyl group of second 3-formylchromone and resulted in formation of an adduct 512 which was submitted to dehydration to afford 513 and subsequently the final product (Scheme 133).
 |
| | Scheme 132 | |
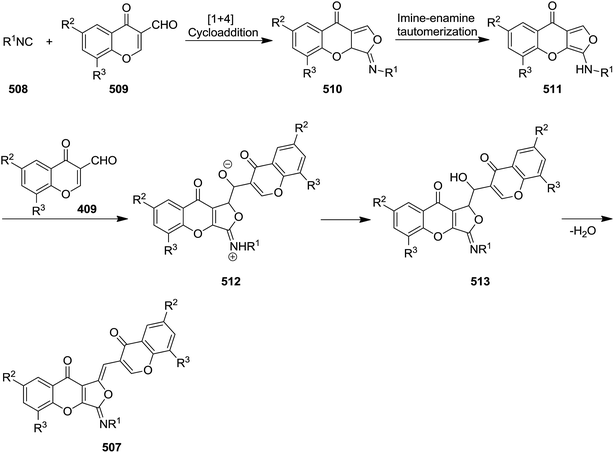 |
| | Scheme 133 | |
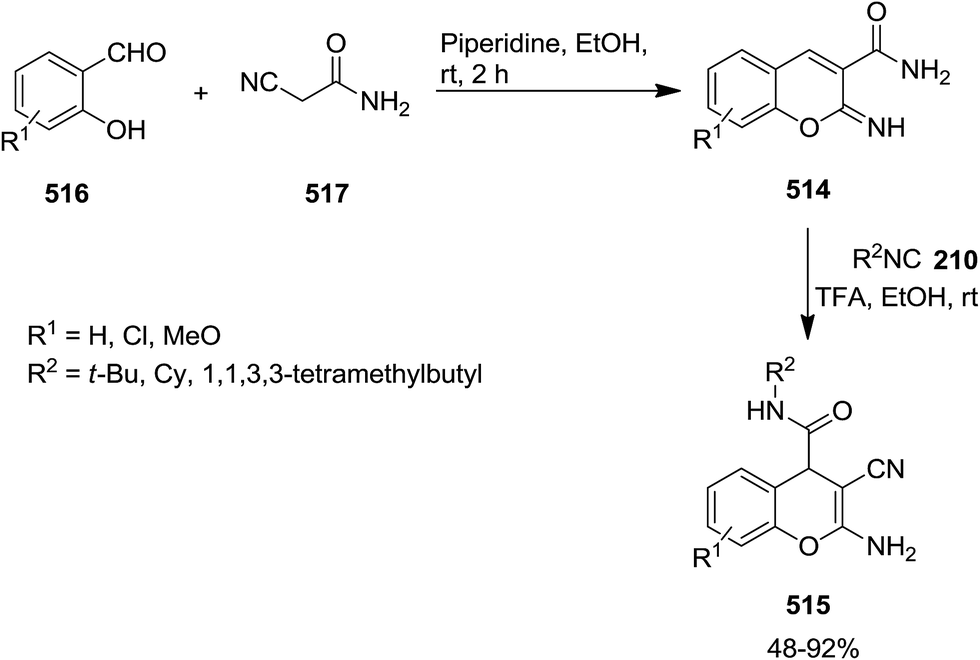
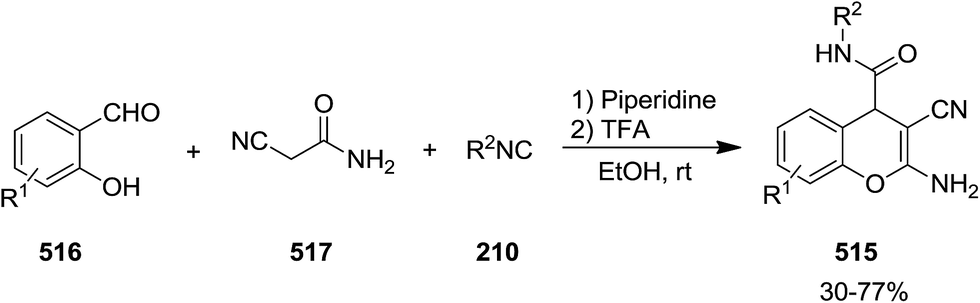
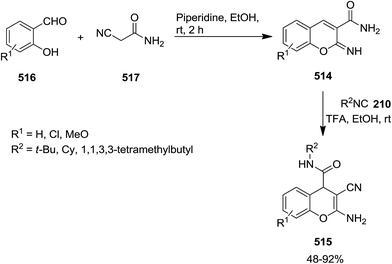
Acid-promoted domino reaction of 2-imino-2H-chromene-3-carboxamide derivatives 514 and isocyanides 210 was utilized for the synthesis of novel 2-amino-3-cyano-4H-chromene-4-carboxamides 515 in high yields (Schemes 134 and 135).242 514 derivatives were obtained via the reaction of salicylaldehydes 516 and cyanoacetamide 517 in the presence of piperidine as catalyst. One pot three-component reaction for the synthesis of 2-amino-3-cyano-4H-chromene-4-carboxamides 515 has also been developed. Salicylaldehydes reacted with 2-cyanoacetamide under this condition to furnish the corresponding 2-imino-2H-chromene-3-carboxamides. The latter participated in a reaction with isocyanide and trifluoroacetic acid to afford the final product.
 |
| | Scheme 134 | |
 |
| | Scheme 135 | |
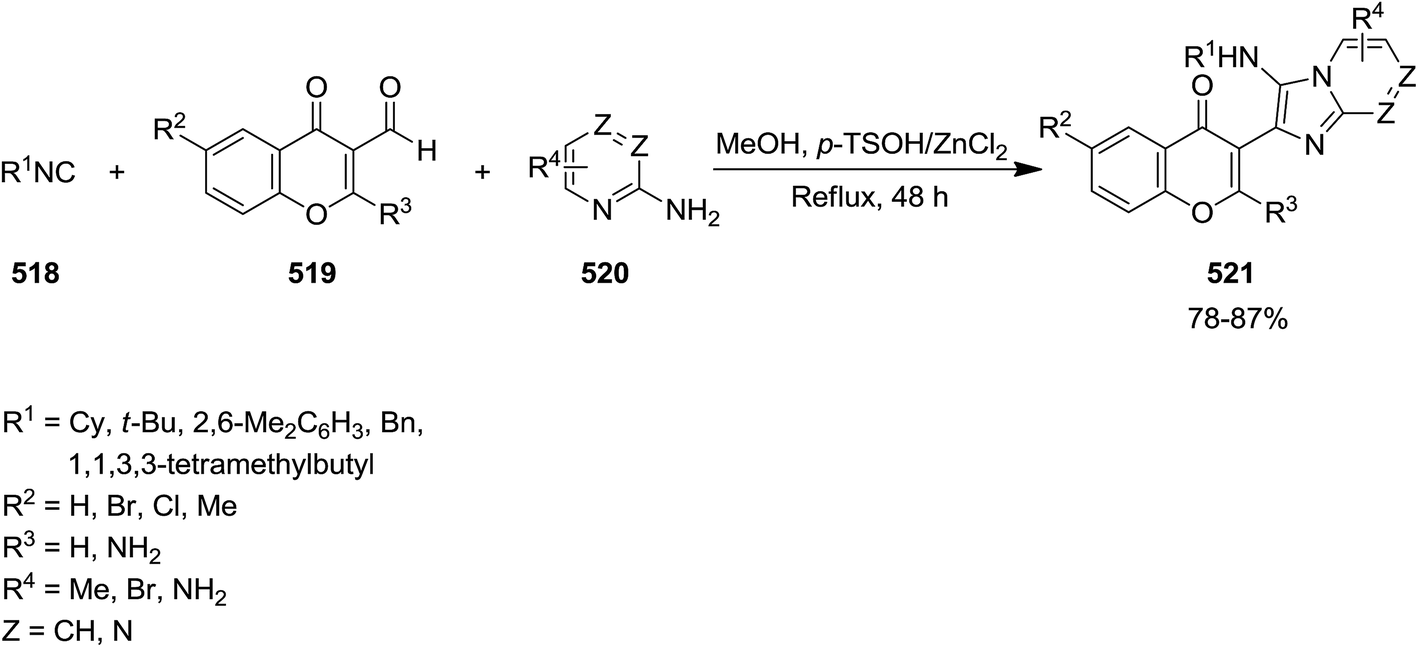
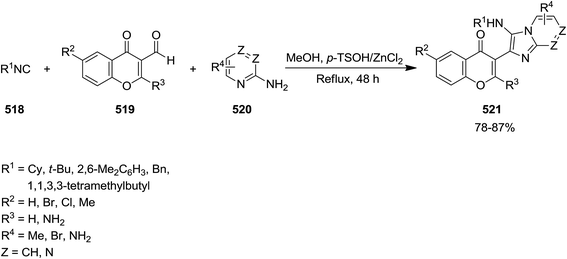
Isocyanide-based synthesis was used for preparing a variety of imidazochromen-4-one derivatives 521 through one-pot reaction of isocyanides 518, 4-oxo-4H-chromene-3-carbaldehydes 519, and 2-aminoazines 520 (Scheme 136).243 The reaction performed under p-TSOH/ZnCl2 catalysis, completed after 48 h refluxing in methanol to afford desired products in high yields. Note worthily, the combination of catalyst was essential for promoting the reaction. The reaction did not proceed in the presence of sole ZnCl2 and led to trace amount of product in the presence of p-TSOH. The procedure showed good functional group tolerance and could be applied for synthesis of compounds with potential biological activities.
 |
| | Scheme 136 | |
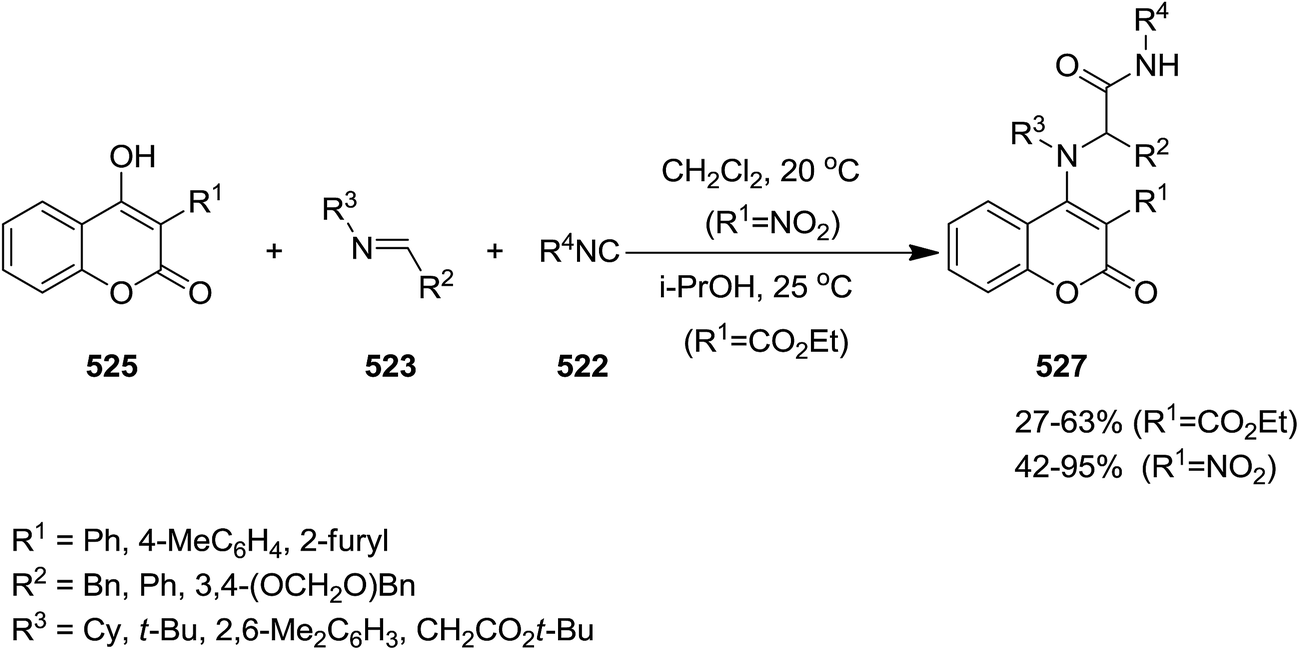
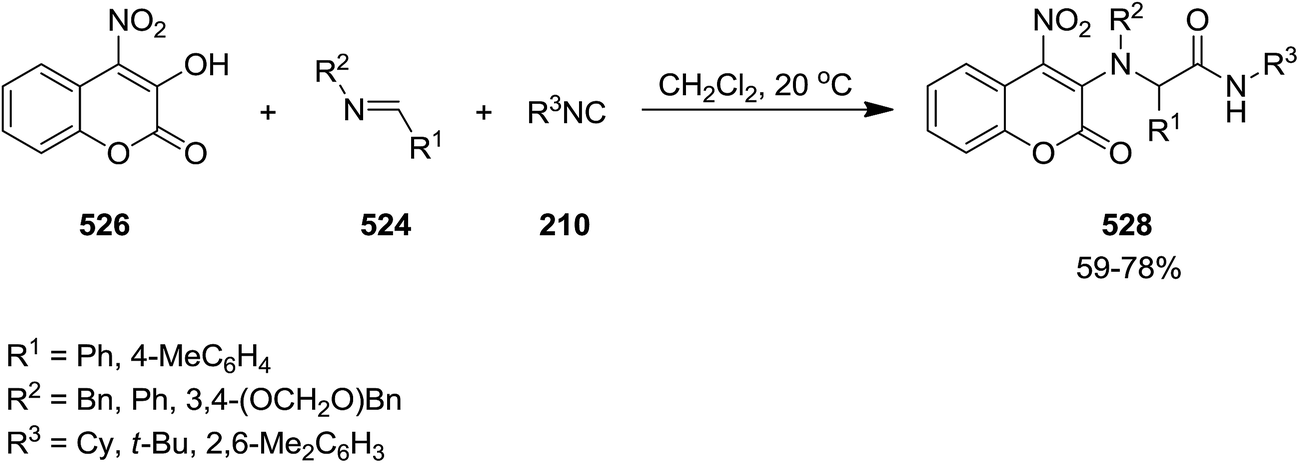
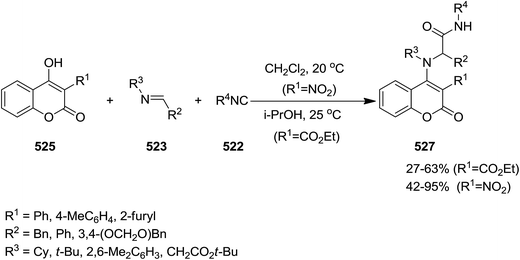
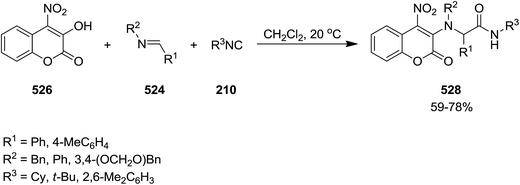
2.3.7 Coumarin. Un-catalyzed, atom-economical three-component enol-Ugi reaction of isocyanides 522 or 239, imines 523 and 524 and electron-deficient hydroxycoumarin 525 and 526 afforded 3- and 4-coumarin enamine derivatives 527 and 528 under mild conditions in moderate to good yields (Schemes 137 and 138).244 The effects of reaction parameters such as temperature, nature of catalyst, electron withdrawing groups at hydroxycoumarin and solvent were also studied. It was found that the reagents were susceptible to decomposition at elevated temperature. Therefore, the reaction was conducted at room temperature. In addition, dichloromethane was found as the solvent of choice.
 |
| | Scheme 137 | |
 |
| | Scheme 138 | |
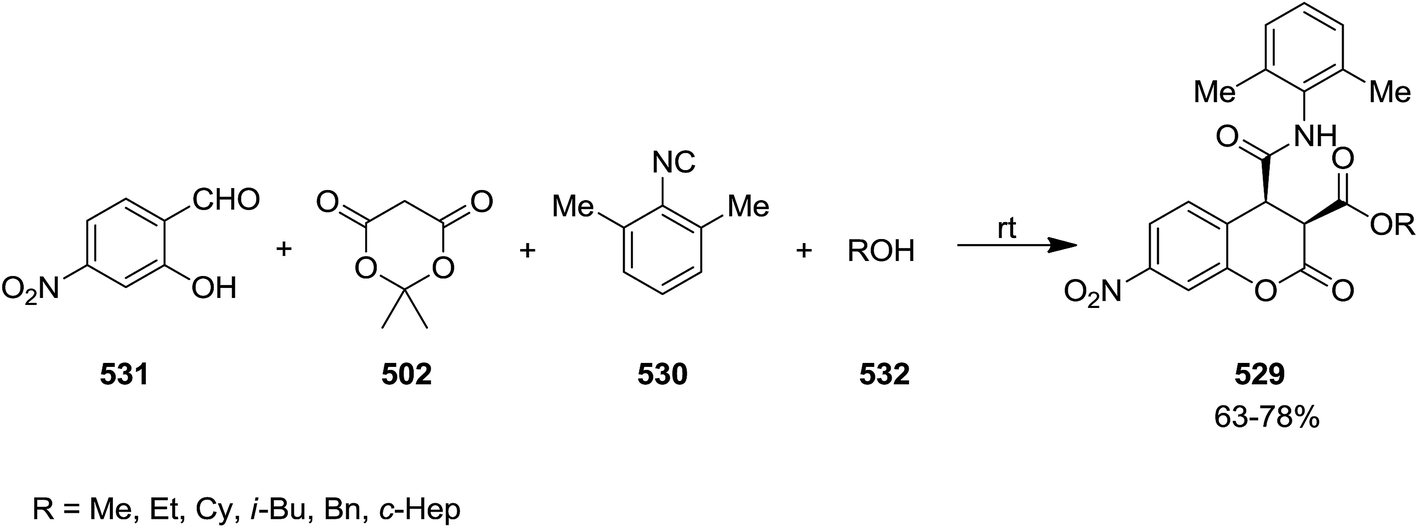
Following isocyanide chemistry, a novel catalyst-free and stereoselective protocol for the synthesis of 3,4-dihydro-7-nitrocoumarins 529 bearing nitro group was reported. This process involves a one pot four-component reaction of 2,6-dimethylphenyl isocyanide 530, 2-hydroxy-4-nitrobenzaldehyde 531, Meldrum's acid 502 and either aromatic or aliphatic alcohols 532 (Scheme 139).245 Mild reaction conditions, good yields, simplicity of work up procedure and short reaction times were the main advantages of this synthetic strategy. Notably, replacing of 2,6-dimethylphenyl isocyanide with 1,1,3,3-tetramethylbutyl isocyanide led to formation of 3,4-dihydrocoumarin derivatives instead of 3,4-dihydro-7-nitrocoumarins.
 |
| | Scheme 139 | |
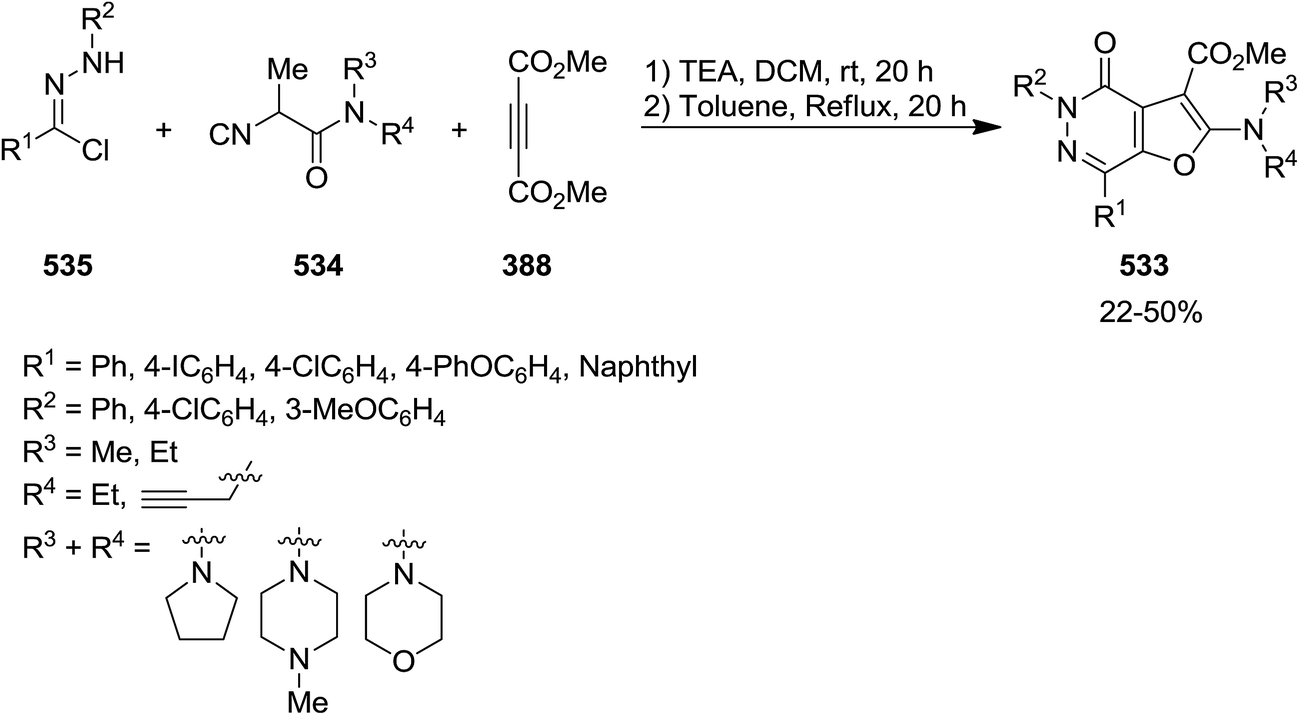
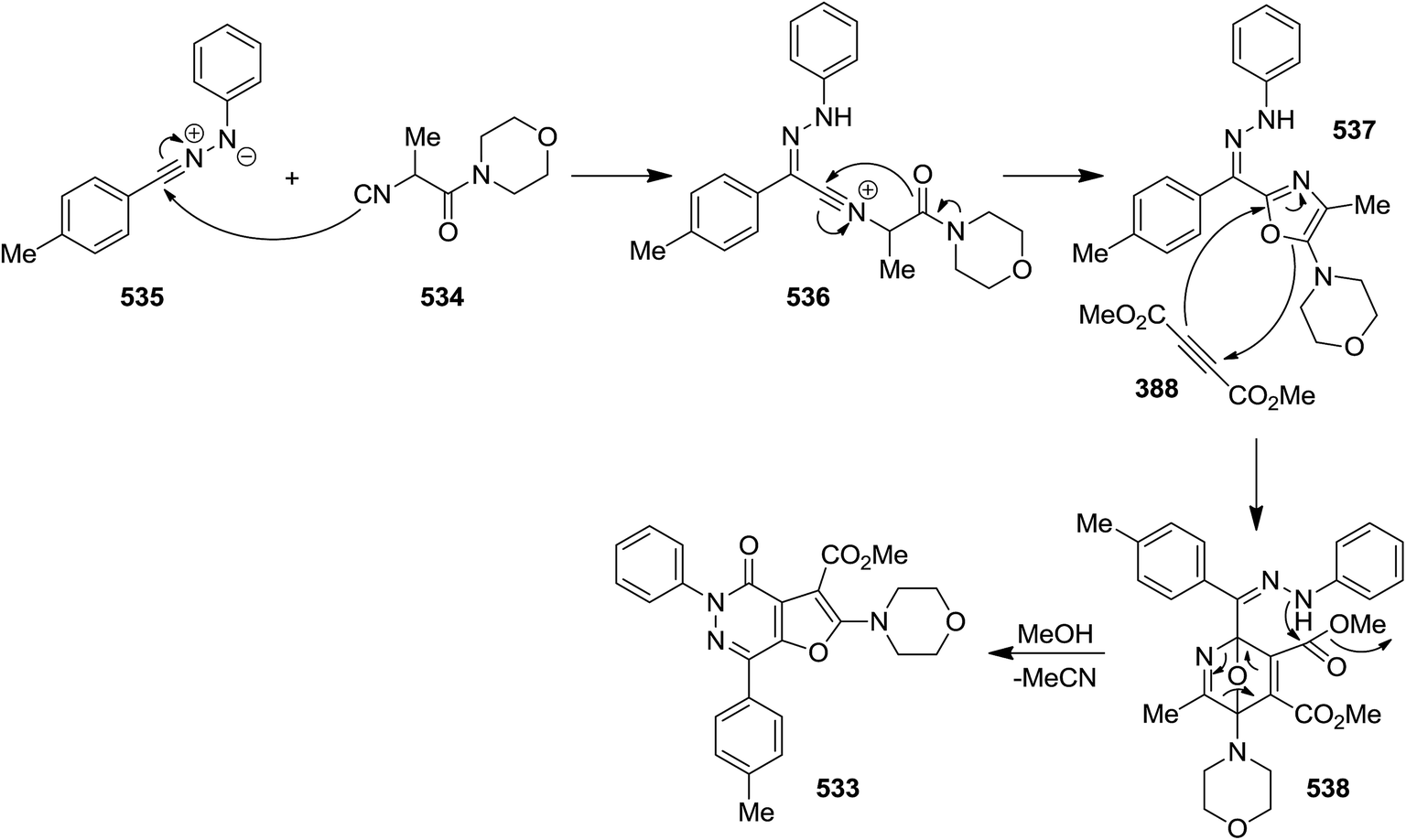
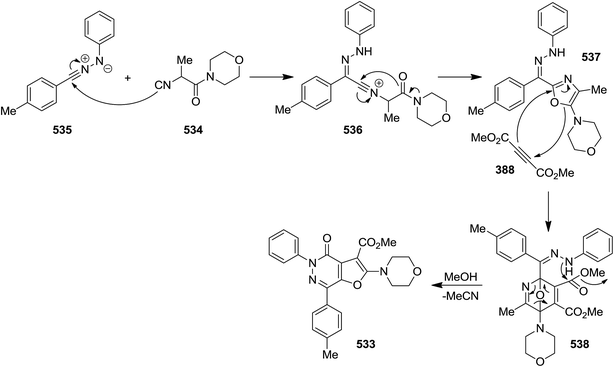
2.3.8 Pyridazin. A novel synthetic protocol was disclosed for the preparation of multi-functionalized furo[2,3-d]pyridazin-4(5H)-one derivatives 533 via a one pot, three-component reaction of hydrazonoyl chlorides 534, isocyanoacetamides 535 and dimethylacetylene dicarboxylate 388 (Scheme 140).246 This domino reaction involves a four sequential reactions including, oxazole formation 537 via intermediate 536/Diels–Alder [4 + 2] cycloaddition and formation of 538/cycloreversion, and intramolecular lactamization (Scheme 141) leading to the desired products in moderate yields. The generality of this strategy was confirmed by using various isocyanides and hydrazonal chlorides. It is worthy to mention that the ester group in final product could be readily hydrolyzed to give an acid functionality which could be used for further derivatization.
 |
| | Scheme 140 | |
 |
| | Scheme 141 | |
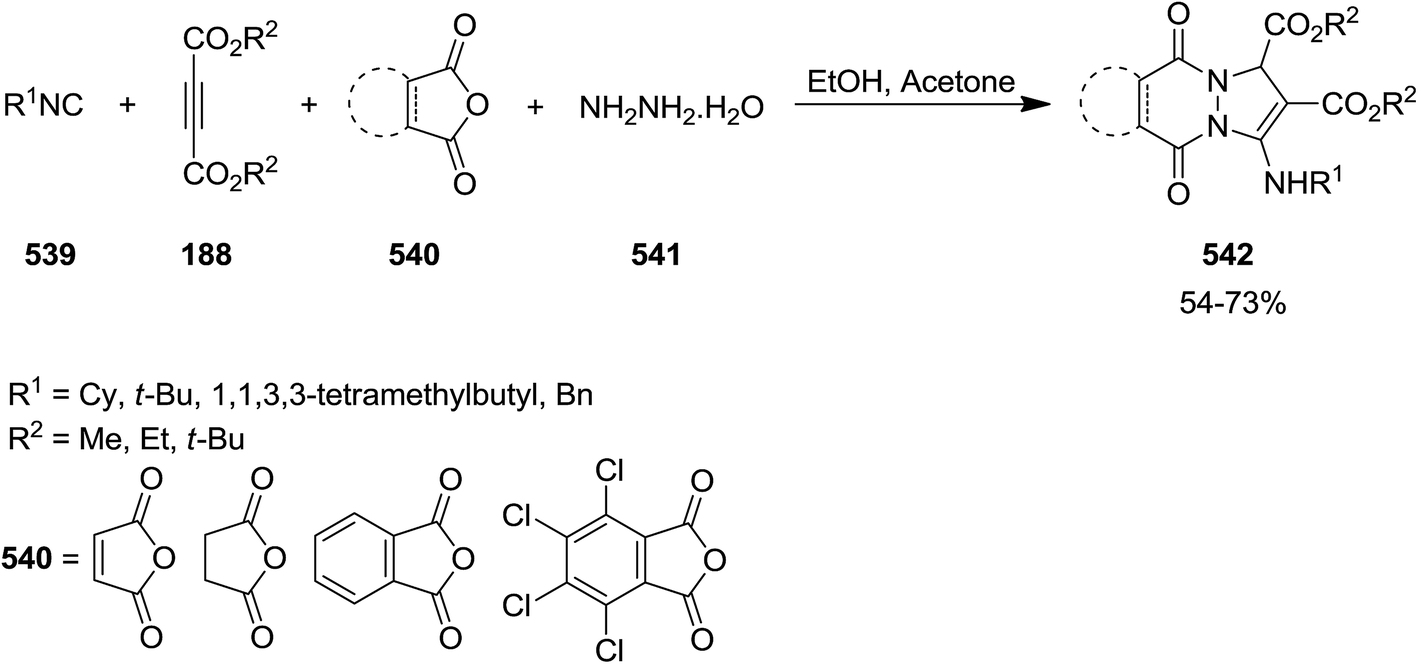
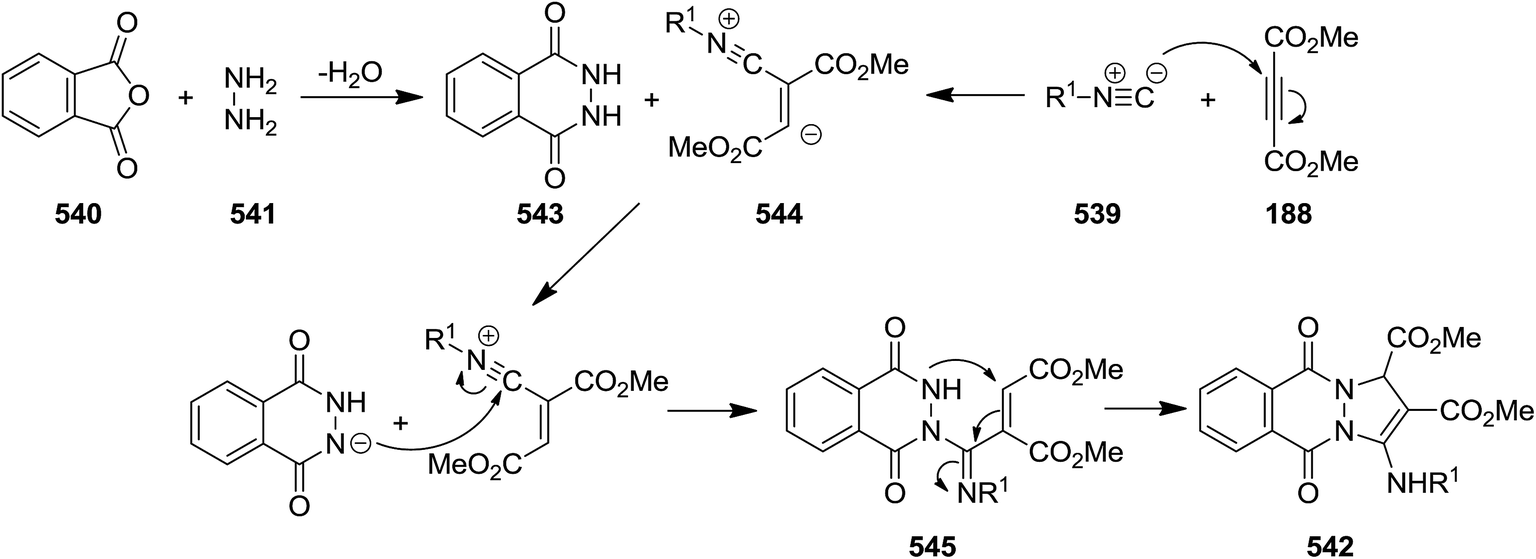
Using four-component reaction of dialkyl acetylenedicarboxylates 188, isocyanides 539 cyclic anhydrides 540, and hydrazine hydrate 541, Shaabani et al. disclosed a new synthetic strategy for synthesis of a series of 1H-pyrazolo[1,2-b]phthalazine-1,2-dicarboxylates and 1H-pyrazolo[1,2-a]pyridazine-1,2-dicarboxylates 542 (Scheme 142).247 Replacing cyclic anhydrides including phthalic anhydride, succinic anhydride and maleic anhydride with phthalimide, isochroman-1,3-dione, N-aminophthalimide and 2-benzofuran-1(3H)-one did not furnish the corresponding products. Mild reaction condition, high yields and broad substrate scope were the advantages of this protocol. The plausible mechanism was reaction of phthalic anhydride 540 and hydrazine 541 to afford 2,3-dihydrophthalazine-1,4-dione 543. Intermediate 544 generated from isocyanide 539 and dialkyl acetylenedicarboxylate 188 would be protonated. Subsequently, the cationic center would be quenched and compound 545 would be formed which transformed to final product via intramolecular nucleophilic reaction (Scheme 143).
 |
| | Scheme 142 | |
 |
| | Scheme 143 | |
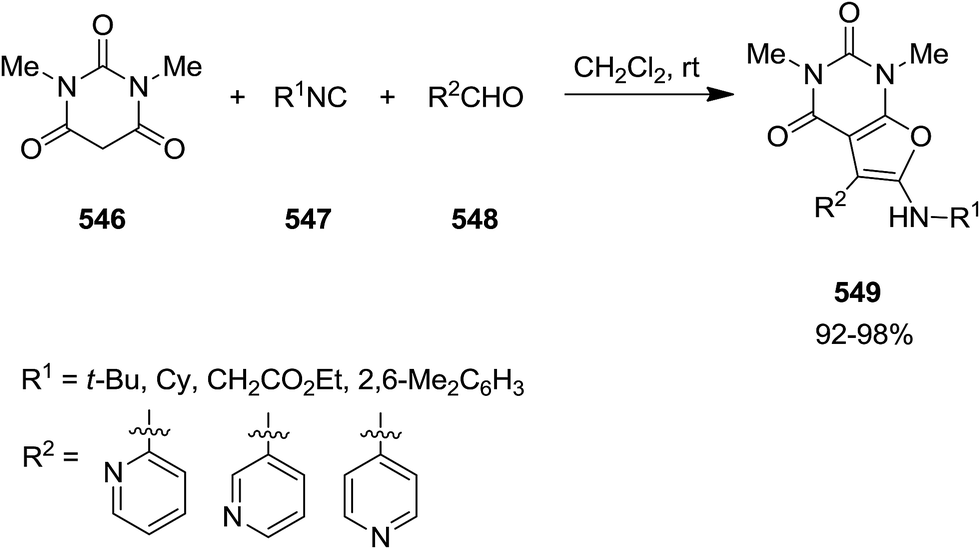
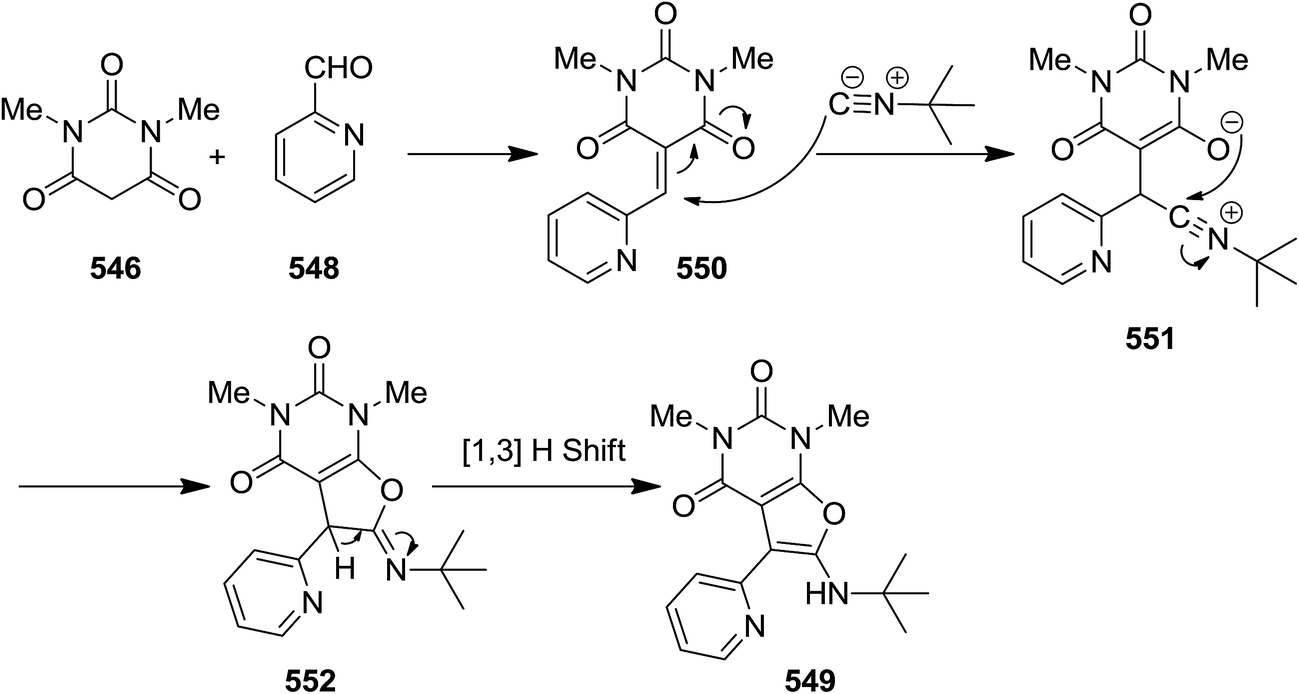
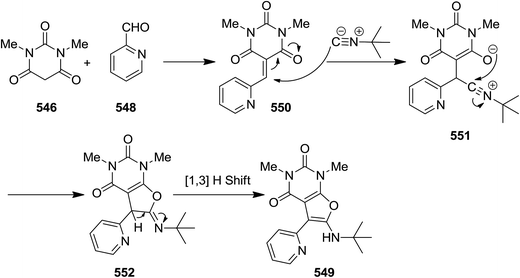
2.3.9 Pyrimidines. A one pot three-component reaction of 1,3-dimethylbarbituric acid 546, alkyl or aryl isocyanides 547 and pyridinecarbaldehydes 548, under mild reaction conditions afforded 5-ryl-6-(alkyl- or aryl-amino)-1,3-dimethylfuro[2,3-d]pyrimidines 549 (Scheme 144).248 It has been proposed that an intermediate 550 generated via a Knoevenagel condensation of 1,3-dimethylbarbituric acid and aldehyde was attacked by an appropriate isocyanide. Subsequently, the adduct 551 of former reaction was submitted to intra nucleophilic addition by oxanion to form a five-membered ring 552. The tautomerization of the latter resulted in the final compound (Scheme 145).
 |
| | Scheme 144 | |
 |
| | Scheme 145 | |
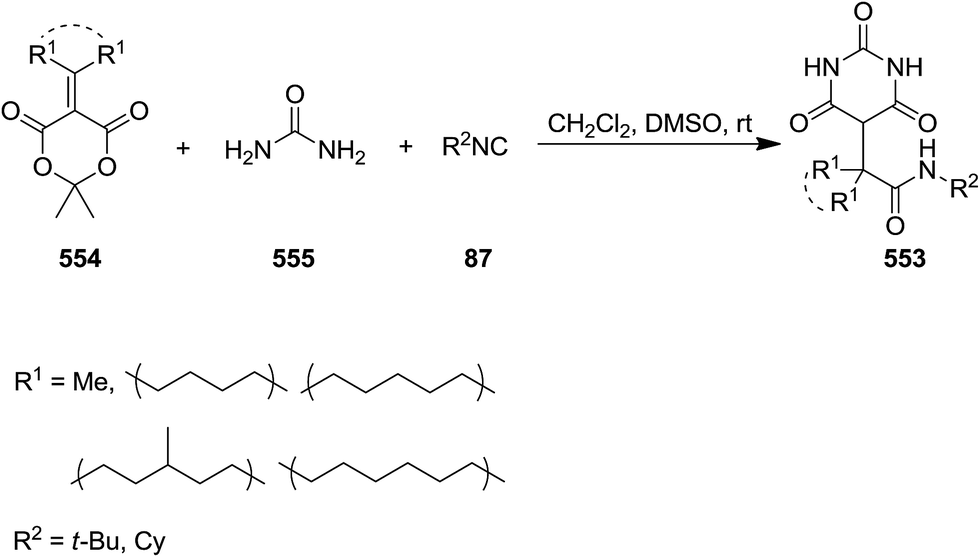
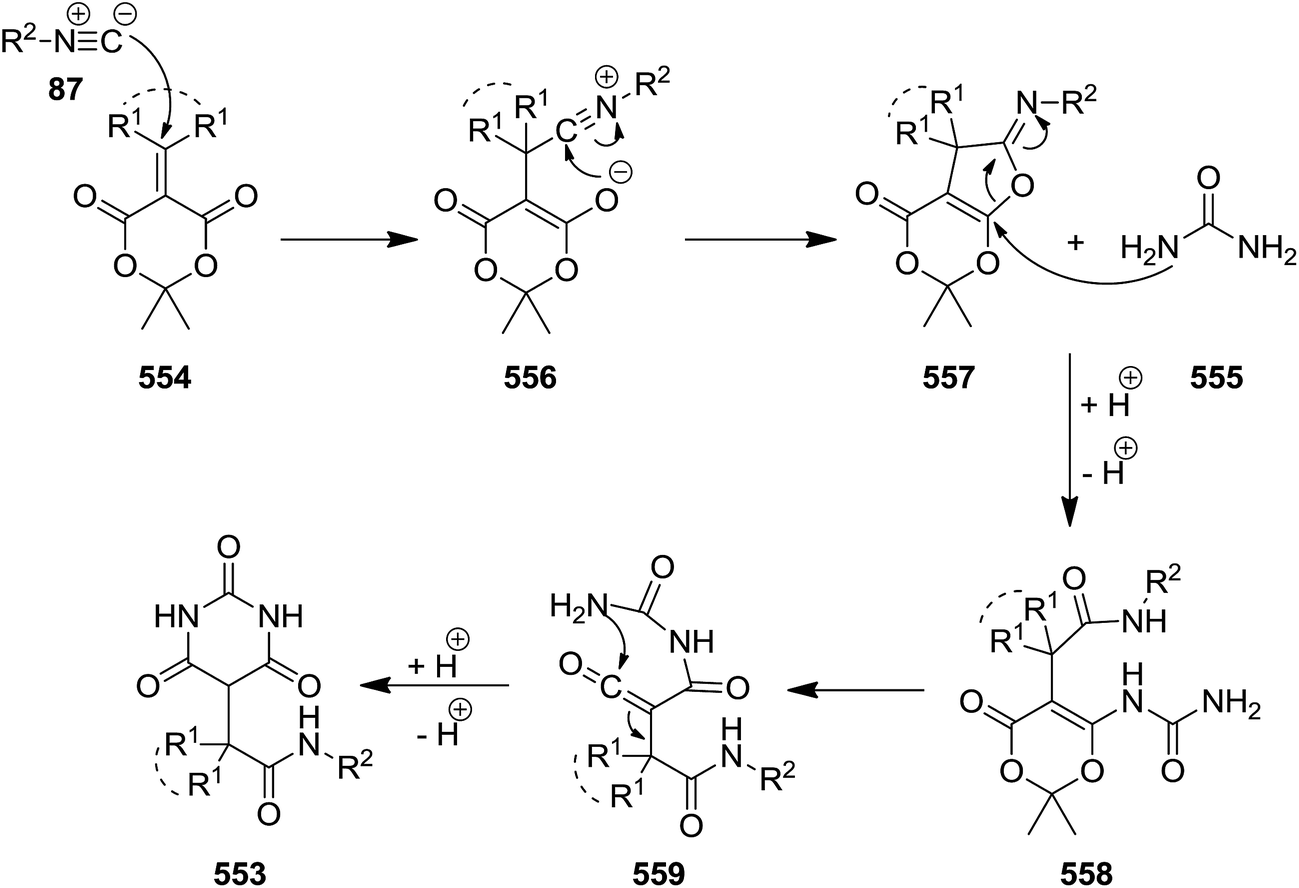
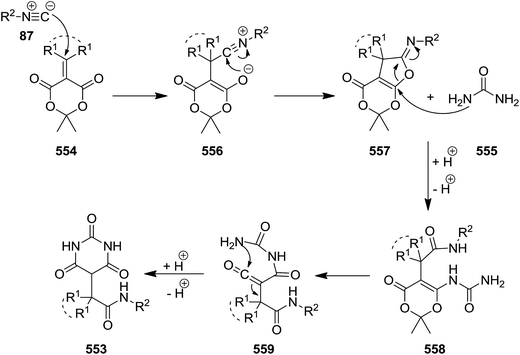
Functionalized barbituric acid derivatives 553 were synthesized via a one-pot three-component reaction of alkyl isocyanides 87, alkylidene-substituted Meldrum's acid 554 and urea 555 under mild reaction conditions, i.e. at room temperature in DMSO/CH2Cl2 (Scheme 146).249 The suggested reaction mechanism involves the formation of an iminolactone intermediate 557 via [4 + 1] cycloaddition of isocyanide and Meldrum's acid 556 followed by conjugate addition of urea and five-membered ring opening to generate a new intermediate 558. Subsequent removal of acetone resulted in the formation of a ketene 559. Nucleophilic attack of urea using its second nitrogen atom to the ketene intermediate furnished the desired product (Scheme 147).
 |
| | Scheme 146 | |
 |
| | Scheme 147 | |
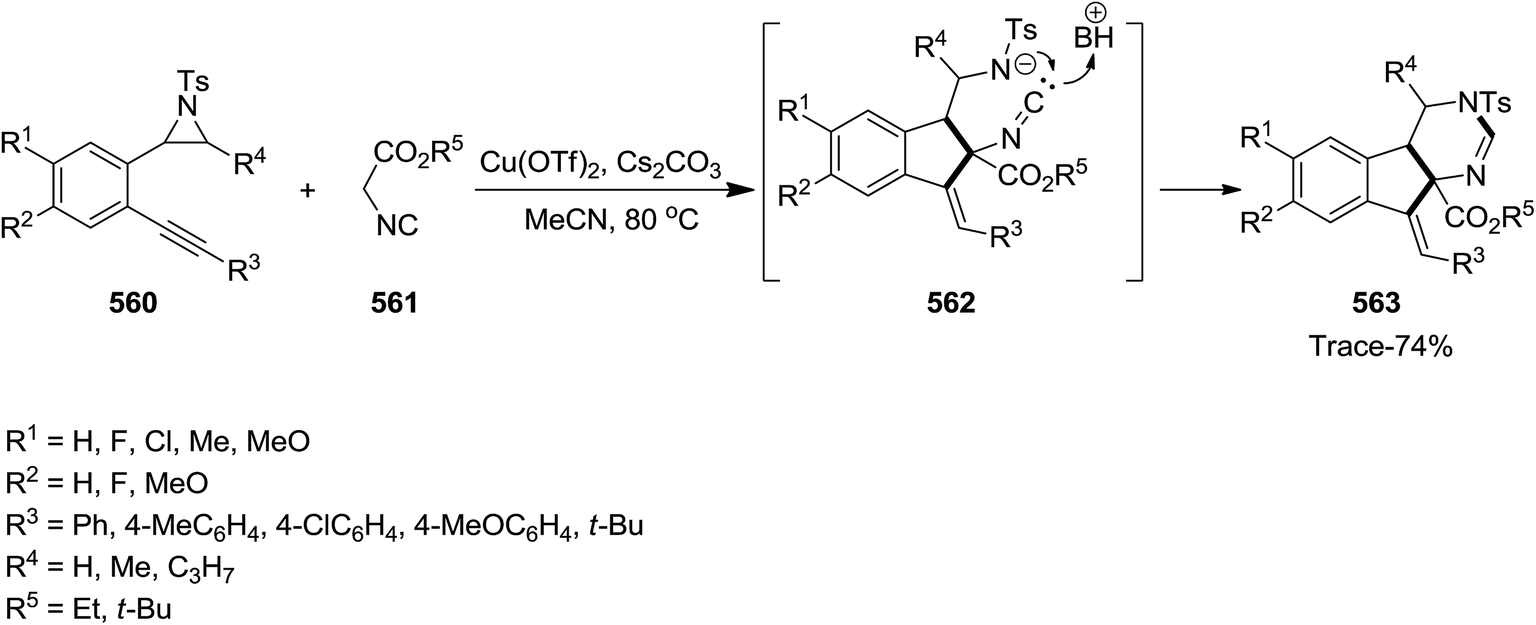
Copper(II)triflate-promoted cascade reaction of 2-(2-alkynylphenyl)-aziridines 560 and 2-isocyanoacetates 561 proceeded in the presence of cesium carbonate as base in acetonitrile to afford tetrahydro-3H-indeno[2,1-d]pyrimidine derivatives 563 in moderate to good yields.250 The proposed mechanism involves the formation of 1-methyleneindene 562 ring followed by reaction of in situ formed nitrogen anion with isocyanide (Scheme 148). The importance of this synthetic strategy lies in the fact that three new bonds as well as two new six and five member rings are generated via a tandem reaction.
 |
| | Scheme 148 | |
Un-catalyzed, one pot three-component reaction of dialkyl acetylenedicarboxylates 564, isocyanides 210 and 2-imino-1,3-thiazolidin-4-one 565 at room temperature in CH2Cl2, affords the biologically attractive highly functionalized 3-oxo-2,3-dihydro-5H-[1,3]thiazolo[3,2-a]pyrimidine derivatives 566 in satisfactory yields (Scheme 149).251 The reaction mechanism consisted of reaction of dialkyl acetylenedicarboxylate and isocyanide to afford a zwitterionic intermediate 567 followed by its protonation by 2-imino-1,3-thiazolidin-4-one and formation of 568. The obtained nitrilium ion was attacked by the conjugate base of the NH-acid 569 to generate a ketenimine 570 as an intermediate. The latter was then subjected into intramolecular annulation to furnish the final product (Scheme 150).
 |
| | Scheme 149 | |
 |
| | Scheme 150 | |
Esmaeili et al. reported a novel protocol for efficient synthesis of tricyclic fused pyranothiazolopyrimidine derivatives 573 trough one-pot multi-component reaction of dialkylacetylenedicarboxylate (DAAD) 571, isocyanide 210 and 2,3-dihydro-5H-[1,3]thiazolo[3,2-a]pyrimidine-5,7(6H)-dione 572 (Scheme 151).252 The proposed mechanism was based on reaction of DMAD 571 and isocyanide 210 and formation of the zwitterion 574. The latter would be protonated to afford a positively charged nitrilium ion 575 which tolerated attack by conjugate base of 572 to furnish 576. The final product would be obtained through tautomerization of the ketenimine to anolate followed by intramolecular cyclization (Scheme 152).
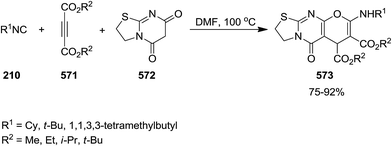 |
| | Scheme 151 | |
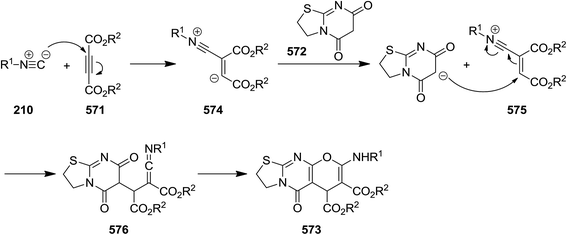 |
| | Scheme 152 | |
2.3.11 Piperazine/phenazine. Bioactive tetrazole-fused ketopiperazine derivatives 596 were obtained via a two-step synthetic strategy. A one pot four-component Ugi reaction of aldehydes 590, isocyanides 591, tritylamine 592 and azidotrimethylsilane 330 gave α-aminotetrazoles 594 in high yields in the first step via intermediate 593 (Scheme 156). The latter subsequently underwent classic intramolecular Ugi reaction with aldehyde 595 and isocyanide 596 under microwave irradiation in trifluoroethanol to furnish the final product 597 in short reaction time (30 min) and moderate yields (Scheme 157).255
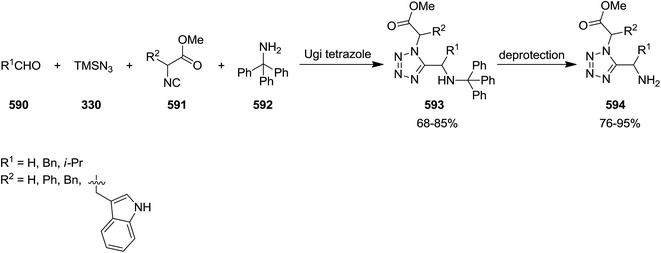 |
| | Scheme 156 | |
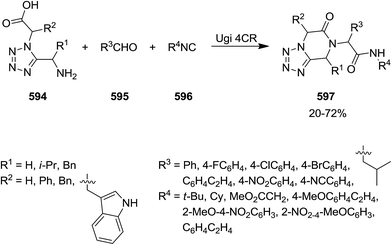 |
| | Scheme 157 | |
A one pot four-component Ugi reaction of isocyanide 87, 2-formylindole 598, bromoacetic acid 218 and amines 599 in methanol led to the formation of an Ugi-adduct 600 which could undergo annulations under basic conditions in DMF to afford 1,2-dihydropyrazino[1,2-a]indol-3(4H)-ones 601 and piperazin-2-ones 602 as major and minor products respectively (Scheme 158). It was found that strong base, such as Cs2CO3, could effectively promote the cyclization while weak base such as K2CO3, NaHCO3 or Na2CO3 did not furnish the desired product even after long reaction time.256
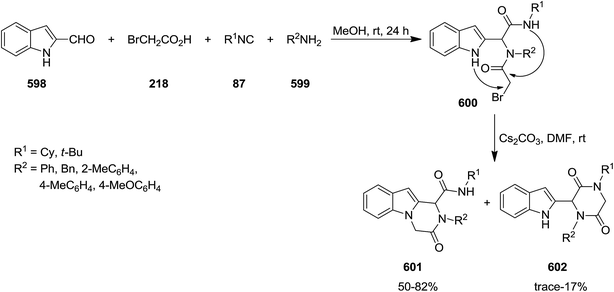 |
| | Scheme 158 | |
Cai et al. developed a new protocol for preparation of 2,5-diketopiperazin 608 through initial synthesis of 2-isocyanophenyl 4-methylbenzoate 607 from Ugi four-component reaction of amines 604 and 606, isocyanides 603 and aldehydes 605 to afford 2-isocyanophenyl 4-methylbenzoate as stable isocyanide. Subsequent deprotection + activation/cyclization led to final product (Scheme 159).257 Besides moderate to good yields and good functional group tolerance, this protocol prevented from using isocyanide with irritating odor.
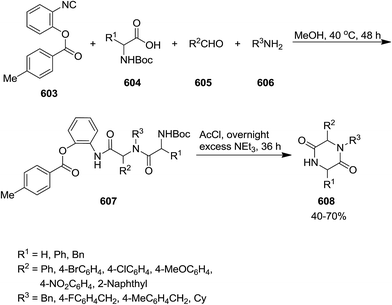 |
| | Scheme 159 | |
Benzo[a]pyrano[2,3-c]phenazine derivatives 611 which possess attractive biological properties were obtained through multi-component reaction of isocyanide derivatives 210, dialkyl acetylenedicarboxylate 49, o-phenylenediamine 609 and 2-hydroxynaphthalene-1,4-dione 610 in excellent yields (Scheme 160).258 The features of this protocol were simplicity, broad substrate scope, use of easily available substrates, mild reaction condition and concomitant formation of two N–C, one C–O and two C–C bonds.
 |
| | Scheme 160 | |
2.3.12 Quinoxalines. Bazgir et al. developed a facile and atom-economical synthetic protocol for the efficient synthesis of pyrano-pyrido-quinoxalines 614 from reaction of dialkyl acetylenedicarboxylates 188 and pyrido[1,2-a]quinoxaline-trione 612 and isocyanide 613 (Scheme 161). In this one pot three-component reaction, a zwitterionic intermediate 615 is generated via reaction of 188 and 613. This intermediate can be protonated to form 616 and subjected to nucleophilic attack by anion of pyrido[1,2-a]quinoxaline-trione 617 to afford ketenimine intermediate 618 which is subjected to cyclization to furnish the final product (Scheme 162).259 In this process, the formation of two C–O and C–C bonds occurred concomitantly.
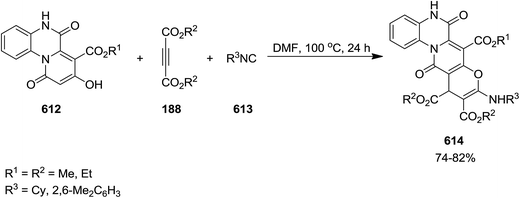 |
| | Scheme 161 | |
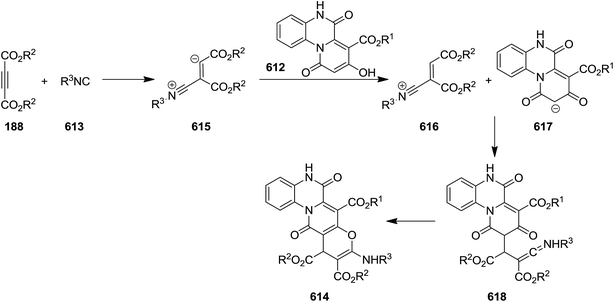 |
| | Scheme 162 | |
Using Pd as a catalyst, a novel synthetic protocol was developed and reported for preparing 2-aminobenzoxazinone derivatives 621 through oxidative coupling reaction of anthranilic acids 619 and isocyanides 620 (Scheme 163). This atom economic reaction proceeded smoothly in mild reaction conditions (O2, 1 atm, and temperature 75–100 °C), and relatively short reaction time (4 h) without generating any hazardous waste (H2O was the only by-product). This reaction showed the practical application of oxidative coupling of isocyanides and bisnucleophiles the synthesis of various heterocycles.260
 |
| | Scheme 163 | |
One-pot multi-component reaction of dialkyl acetylenedicarboxylates 49, isocyanides 87 and 5-substituted uracils 622 under mild reaction conditions, i.e. room temperature in 1,4-dioxane resulted in the formation of biologically attractive multi functional pyrimido[2,1-b][1,3]oxazines 623 in high yields (Scheme 164).261 The reaction started from isocyanide insertion to acetylenic ester and formation of 624 followed by its protonation and formation of 625. The protonated species is attacked by an anion 626 derived from uracil to generate a ketenimine intermediate 627 which is then subjected to tautomerization and cyclization to afford 628 and finally the desired product (Scheme 165).
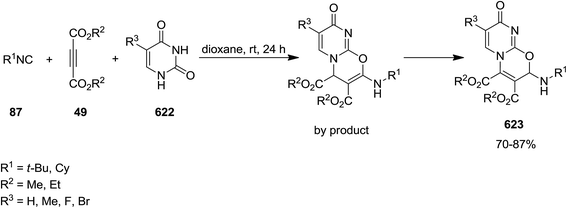 |
| | Scheme 164 | |
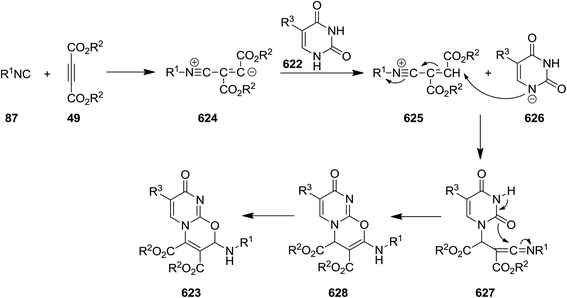 |
| | Scheme 165 | |
Jiang et al. developed a facile, mild and Pd-catalyzed method for the synthesis of 2-aminobenzoxazoles 631 and 3-aminobenzoxazines 632 in good to high yields from reaction of aminophenol 629 and isocyanide 630 (Scheme 166).262 This synthetic strategy could be applied to wide range of substrates and reagents with various functionalities. Testing various types of Pd catalysts in this reaction, indicated that the catalysts had an essential role on the formation of the product. In this line, however Pd(PPh3)2-catalyzed reaction resulted in 2-aminobenzoxazoles while PdCl2 led to the formation of 3-aminobenzoxazines.
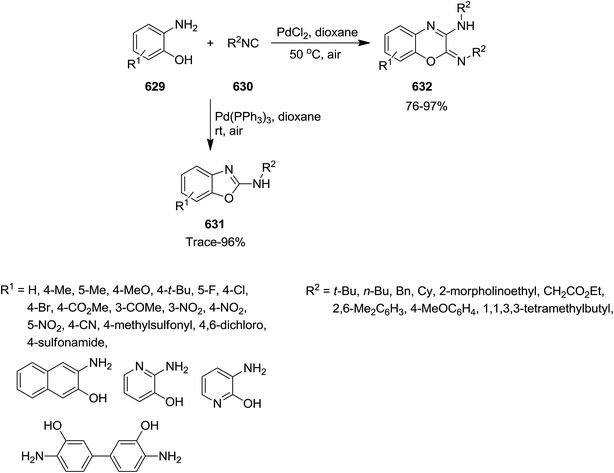 |
| | Scheme 166 | |
2.3.13 Thiazine. Cai et al. promoted an efficient synthetic approach for the synthesis of 5H-benzo[d]imidazo[5,1-b][1,3]thiazine 635 with broad substrate scope.263 This approach was based on cascade reaction of o-alkynylphenyl isothiocyanates 633 with isocyanides 634 (Scheme 167). The effects of reaction variables such as catalyst, solvent and base were studied in order to optimize the reaction conditions. THF, Cs2CO3 and CuCl were selected as the most appropriate solvent, base and catalyst respectively. The possibility of using 2-isothiocyanato-3-(phenylethynyl)pyridine 636 reagent in its reaction with ethyl 2-isocyanoacetate 15 was investigated. The formation of the desired product, i.e. (Z)-ethyl 5-benzylidene-5H-imidazo[5,1-b]pyrido[2,3-d][1,3]-thiazine-7-carboxylate 637 (Scheme 168), proved the broad scope of this approach.
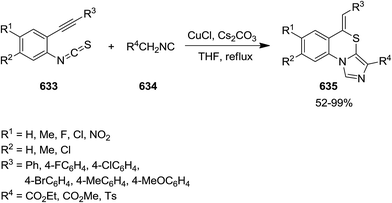 |
| | Scheme 167 | |
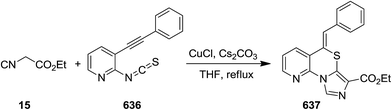 |
| | Scheme 168 | |
Pyrimido[2,1-b][1,3]thiazines 639 with potential use in pharmacology were synthesized through a one pot three-component reaction of thiouracils 638, isocyanides 87 and dialkyl acetylenedicarboxylates 188 in high yields (Scheme 169).264 The reaction mechanism included insertion of isocyanide to dialkyl acetylenedicarboxylate and the formation of a 1:1 zwitterionic intermediate which subsequently reacted with thiouracil to generate a ketenimine intermediate. Finally, the annulations and rearrangement of this intermediate furnished the final product.
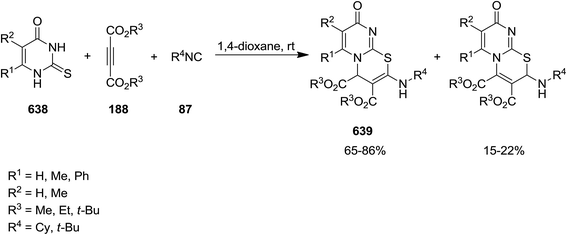 |
| | Scheme 169 | |
Batra et al. developed a novel approach for synthesis of 4-substituted imino-4H-benzo[d] [1,3]thiazin-2-amine derivatives 642 through isocyanide 640 insertion in 2-bromophenylthioureas 641 and cross coupling reaction by the thiol group under Pd catalysis (Scheme 170).265 In the presence of cyclohexyl isocyanide, various aliphatic and aromatic 2-bromophenylthioureas could furnish the desired product in high yields while other isocyanides (except 2-isocyanoacetate) led to corresponding products with only aromatic 2-bromophenylthioureas.
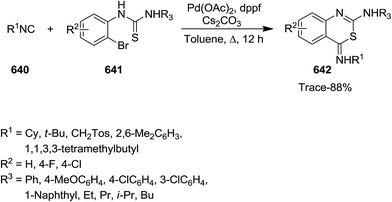 |
| | Scheme 170 | |
2.3.14 Oxadiazin. Based on the well-established Passerini and Ugi reaction mechanism and in attempt to develop intramolecular trapping of the nitrilium intermediate, generated in this reaction, Soeta and Ukaji used a substrate, C,N-cyclic N′-acyl azomethine imines 643, bearing both electrophile and nucleophilic moieties, in [5 + 1] cycloaddition reaction with isocyanides 644 to furnish imin-1,3,4-oxadiazin-6-ones 645 (Scheme 171).266 Mild reaction conditions, wide substrate scope and obviating the need for catalyst were the advantages, claimed for of this synthetic strategy.
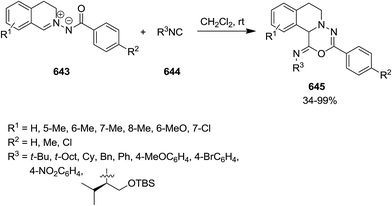 |
| | Scheme 171 | |
2.4. Large membered ring
Bazgir et al. reported a catalyst-free synthesis of a new series of 2-aryl-2-(2,3,4,5-tetrahydro-2,4-dioxo-1H-1,5-benzodiazepin-3-yl)acetamide derivatives 649 under mild reaction condition. In this multi-component synthetic strategy, Meldrum's acid 502, benzene-1,2-diamine 646, aldehydes 647, isocyanides 648 in water reacted to afford the desired product in good yields (Scheme 172).267 The reaction proceeded via the formation of 650 and 1,5-benzodiazepine 651 and its Knoevenagel condensation with aldehyde to afford an intermediate 652. The latter underwent Michael-type addition reaction of an isocyanide to generate a new intermediate 653 and dehydration to form 654 which tautomerizes to afford the final product (Scheme 173). Replacing of the aldehyde with ferrocenecarboxaldehyde led to the formation of ferrocene-containing benzodiazepines.
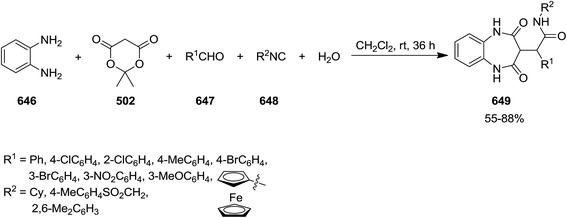 |
| | Scheme 172 | |
 |
| | Scheme 173 | |
Chattopadhyay et al. developed and reported a novel strategy for the synthesis of imidazole-fused benzodiazepinone derivatives 658 in good yields under mild reaction condition via a one pot three-component Ugi-type reaction of anthranilic acids 655, isocyanides 656 and imidazole-2-carbaldehyde 657 (Scheme 174).268 The proposed mechanism was based on attack of the amino group of the anthranilic acid to the carbonyl carbon of the imidazole-2-carbaldehyde to form an imine 658 followed by isocyanide insertion and the formation of an intermediate 659. The latter generated a carboxylate anion which attacked the isocyanide carbon to generate acylated isoamide 660. This compound underwent Mumm rearrangement via acyl transfer to furnish final product (Scheme 175).
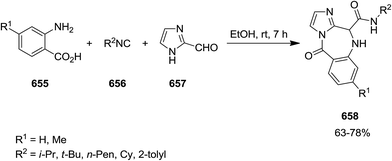 |
| | Scheme 174 | |
 |
| | Scheme 175 | |
Jiang et al. introduced a new synthetic methodology for the synthesis of 4-amine-benzo[b][1,4]oxazepines 664 via multicomponent cascade reaction of isocyanides 661, o-aminophenols 662 and bromoalkynes 663 in high yields, with broad substrate scope (Scheme 176).269 The plausible mechanism for this Pd-catalyzed system involves the formation of an intermediate 665 through reaction of bromoalkyne and o-aminophenol, followed by its oxidative addition to Pd(0) species 666 and formation of a vinyl palladium species. There after a new intermediate 667 is generated via migratory insertion of isocyanide. Subsequently an eight membered azapalladacyclic intermediate 668 is formed via base-assisted removal of hydrogen bromide. Reductive elimination produced an appropriate intermediate 669, which can be isomerized to afford the final product (Scheme 177).
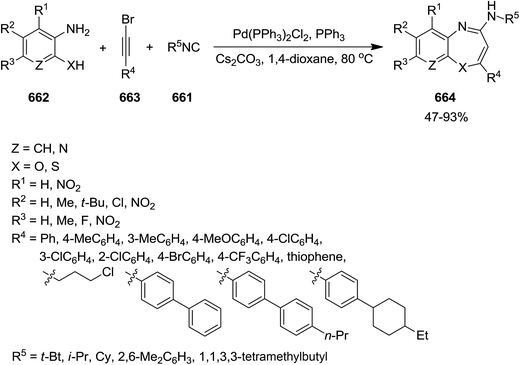 |
| | Scheme 176 | |
 |
| | Scheme 177 | |
Ghandi et al. synthesized a series of novel benzimidazole-fused 1,4-diazepine-5-one derivatives 673 through Ugi 4-center-3-component of alkyl isocyanides 670, amines 671 and 3-(2-formyl-1H-benzimidazol-1-yl)propanoic acid 672 in moderate to high yields (Scheme 178). The latter was prepared via a four step process.270 The generality of the process was confirmed by using various isocyanide and amine derivatives.
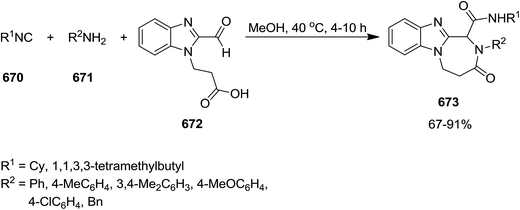 |
| | Scheme 178 | |
Chattopadhyay et al. reported synthesis of new eight-membered 1,5-benzodiazocine-2-one derivatives 678 through a one-pot, two-step synthetic procedure. In the first step, Ugi four-center three-component reaction of aromatic nitro aldehydes 675, isocyanides 674 and β-alanine 676 resulted in formation of β-lactams 677 which subsequently tolerated reductive cyclization in the presence of Fe/NH4Cl (Scheme 179).271 It is worth mentioning that using o-tolyl and 2-naphthyl isocyanides did not furnish the desired products in high yields and only aliphatic isocyanides were suitable for this process.
 |
| | Scheme 179 | |
Novel thioester isocyanide derivatives 679 were obtained from α-amino acids (Scheme 180) and subsequently employed for the synthesis of peptide macrocycles 682 from macrocyclization unsubstituted aziridine aldehyde 680 and peptide 681 (Scheme 181).272 The reaction variables such as solvent, temperature and concentration were optimized. The best solvent recognized was hexafluoroisopropanol. It was found that the temperature did not affect the reaction in terms of reaction rate and yields of the products. Furthermore, side chain conjugation was performed in a chemoselective manner with compound 683 to develop cycle-tail peptides 684 with potential activity against Gram-negative pathogens.
 |
| | Scheme 180 | |
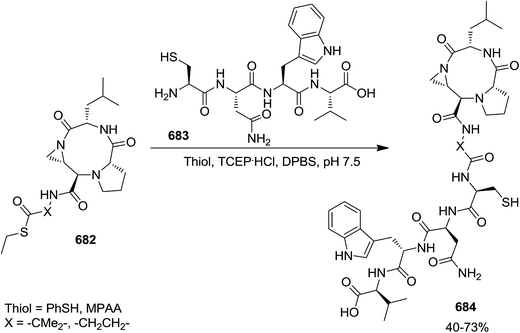 |
| | Scheme 181 | |
Microwave-assisted, one pot multi-component reaction of amino alcohols 685, isocyanides 686 and acid–aldehydes 687, was achieved and reported by Sheppard et al. They developed a novel, efficient and rapid synthetic strategy to obtain medium to large membered lactones 688 (Scheme 182).273 In the case of use of L-prolinol as chiral amino alcohol, diastereomeric eight membered lactones 689a and 689b were furnished in a 1.5:1 ratio which could be separated simply by chromatography (Scheme 183).
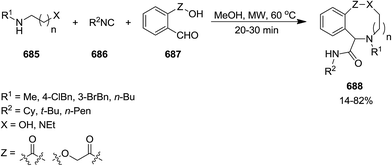 |
| | Scheme 182 | |
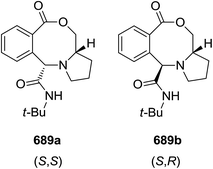 |
| | Scheme 183 | |
Using azidobenzaldehydes 690 as starting compounds, triazolofused benzoxazepinones 694 were successfully synthesized via a two-step reaction. Initially, Passerini reaction of aromatic azidoaldehydes 690, isocyanide 691 and propynoic acids 692 in dichloromethane at room temperature led to the formation of Passerini-adduct 693. It is worth noting that in this low temperature the possibility of reaction of acetylene with isocyanide would be decreased and the desired adducts could be obtained in moderate to good yields. The second step for obtaining desired heterocycle could be performed under either reflux condition or microwave irradiation. The microwave-assisted method could not be generalized to all derivatives. Furthermore, it led to the formation of undesired by-products which made purification inevitable. Post-condensation under reflux condition, however, was more reliable and cleaner (Scheme 184). To afford triazolo-fused benzoxazepines 694, 2-azidobenzaldehydes 690, acetic acid 695 and isocyanides 691 reacted to furnish Passerini-adduct 696. The latter underwent hydrolysis/alkylation with 697 to produce 698. The final products were obtained under microwave irradiation (Scheme 184).274
 |
| | Scheme 184 | |
2.5. Spiro heterocycles
The efficient, atom-economical and facile synthesis of various derivatives of bioactive azaspiro[4.5]trienone 702 was reported by Kumar Srivastava et al.275 The synthetic rout involved cascade four-component Ugi reaction and iodine-mediated iso-iodocyclization (Scheme 154). The Ugi reaction consisted of isocyanides 87, aldehydes 699, p-anisidines 700 and 3-alkyl/aryl-propiolic acids 701. This investigation was extended to the utility of the obtained products for deiodination reaction (Scheme 185) and Pd-catalyzed Suzuki cross coupling reaction (Scheme 186) to afford modified and poly-functionalized azaspiro[4.5]trienones 703 and 704 in good to excellent yields.
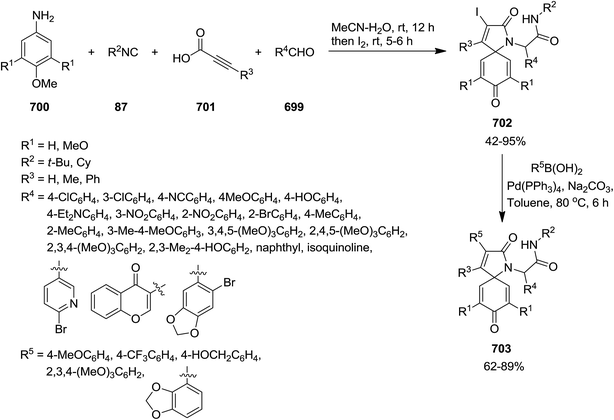 |
| | Scheme 185 | |
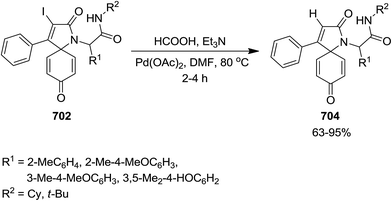 |
| | Scheme 186 | |
Tetrabutylammonium hydrogensulfate (TBAHS)-promoted synthesis of spiroiminolactones 707 was achieved via a one pot multi-component reaction of 1,3-dione derivatives 705, isocyanides 210 and reactive cyclic carbonyl compounds 706 in aqueous media (Scheme 187).276 Simplicity of work up, high yields, the use of environmentally-benign solvent, broad substrate scope and high atom economy were the merits of this synthetic strategy. It is worth nothing that the catalytic amount (10 mol%) of catalyst was essential for the reaction to proceed smoothly and the reaction in the absence of catalyst led to negligible conversion. Furthermore, using higher amount of catalyst did not affect the yield of the desired product. The catalyst also has zwitterionic solubility character and can easily be diffused from the aqueous phase to the aggregated organic phase and vice versa. Another effective factor in this process was the utilization of water as solvent. The water molecules could stabilize the transition state via the formation of hydrogen bonding.
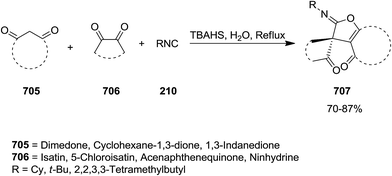 |
| | Scheme 187 | |
Soleimani et al. developed a novel and atom economic strategy for efficient synthesis of 1,7-diazaspiro[4,4]nonane-2,6-dione derivatives 711 (ref. 277) through multi-component reaction of malononitrile 476, primary alkyl amines 710, isocyanides 210 and 2-formylbenzoic acid 708 (Scheme 188). The authors investigated the effect of solvent and demonstrated that the four component reaction in ethanol led to formation of 2-(1-(alkylcarbamoyl)-2,2-dicyanoethyl)-N-alkylbenzamide 712 while 1,7-diazaspiro[4,4]nonane-2,6-diones 711 would be the major product by performing the reaction in dichloromethane and addition of primary alkyl amine after reaction of three other components and formation of isochromeno[3,4-b]pyrroles 709.
 |
| | Scheme 188 | |
2.6. Miscellaneous
Baharfar et al. disclosed a isocyanide-based strategy to obtain highly substituted ketenimines 714 and bis(ketenimines) 716 and 717 via a one-pot, three-component reaction of dialkyl acetylenedicarboxylates 188, pyridine-2-carboxaldoxime 713 or α-furildioxime 715 and alkyl isocyanides 87 or 117 (Schemes 189 and 190). The proposed mechanism was based on isocyanide insertion to dialkyl acetylenedicarboxylate and the generation of an intermediate 718 which subsequently is easily protonated to form 719. The desired product could be obtained via attack of the conjugate base 720 of the OH-acid to the positively charged species (Scheme 191).278
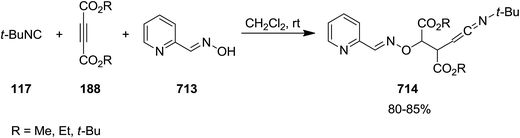 |
| | Scheme 189 | |
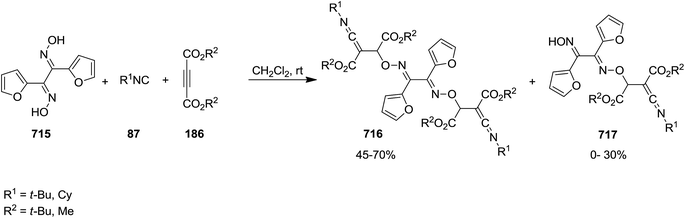 |
| | Scheme 190 | |
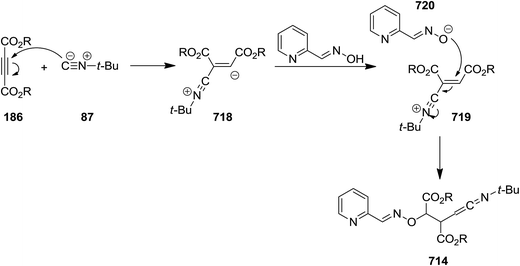 |
| | Scheme 191 | |
Poly-substituted 4-aminoxanthone derivatives 728 were obtained in good to excellent yields via an un-catalyzed one-pot three component reaction of isocyanides 726, 3-carbonylchromones 725 and appropriate dienophiles 727 (Scheme 192).279 For the preparation of chromone 725, initially, condensation of ortho-hydroxyacetophenones 721 with dimethylformamide dimethyl acetal 722 was applied to generate enaminones 723. The latter was reacted with methyl chlorooxoacetate 724 and pyridine under microwave irradiation to furnish the corresponding chromenones. Mild reaction condition, broad substrate scope and simplicity of workup were the advantages of this synthetic strategy.
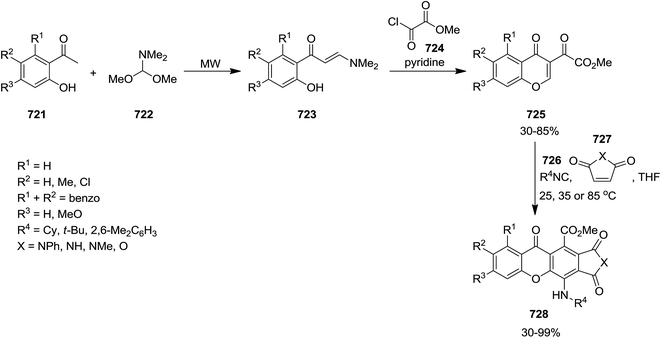 |
| | Scheme 192 | |
Ruiz et al. disclosed synthesis of azaphosphaheterocycles 732 through one-pot multi-component reaction of isocyanides 729, diphosphinoketenimines 730 and water or ethanol 731 (Scheme 193).280 The authors proposed a mechanism based on formation of adduct 733 via α-addition of 730 to isocyanide 729. Subsequently, adduct 733 tolerated oxidative addition at the exocyclic diphenylphosphino group to afford 734. It is worth noting that the presence of water or ethanol is essential for this process. Compound 735 would be formed via transfer of P–H hydrogen to C atom of the former isocyanide. The H transfer furnished compound 736 which subsequently gave the final product (Scheme 194).
 |
| | Scheme 193 | |
 |
| | Scheme 194 | |
3. Conclusion
In this review article, in order to update our previous publication, we tried to highlight the recent applications of isocyanides for the synthesis of diverse heterocycles with various type and numbers of hetero atoms and ring size. As discussed, isocyanide is an attractive reagent which can participate in one-step, cascade or domino synthetic processes for the preparation of a wide range and different size of heterocyclic systems with various functional groups. We hope this review article provide more insight into isocyanide chemistry and encourage readers to take part in developing this extensive filed.
Acknowledgements
MMH is thankful to Iran National Science Foundation (INSF) for partial financial assistance. The authors appreciate partial financial supports from Alzahra University and Iran Polymer and Petrochemical Institute.
References
- S. Sharma, R. A. Maurya, K.-I. Min, G.-Y. Jeong and D.-P. Kim, Angew. Chem., Int. Ed., 2013, 52, 7564–7568 CrossRef CAS PubMed.
- J. G. Rudick, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3985–3991 CrossRef CAS.
- L. Kou, M.-J. Wang, L.-T. Wang, X.-B. Zhao, X. Nan, L. Yang, Y.-Q. Liu, S. L. Morris-Natschke and K.-H. Lee, Eur. J. Med. Chem., 2014, 75, 282–288 CrossRef CAS PubMed.
- A. Sehlinger, B. Verbraeken, M. A. R. Meier and R. Hoogenboom, Polym. Chem., 2015, 6, 3828–3836 RSC.
- H. Duan, Z. Chen, L. Han, Y. Feng, Y. Zhu and S. Yang, Org. Biomol. Chem., 2015, 13, 6782–6788 CAS.
- R. Wang, H. Jianga, Y. Cheng, A. A. Kadi, H.-K. Fun, Y. Zhang and S. Yu, Synthesis, 2014, 46, 2711–2726 CrossRef CAS.
- G. Qiu, Q. Ding and J. Wu, Chem. Soc. Rev., 2013, 42, 5257–5269 RSC.
- L.-R. Wen, M.-C. Lan, W.-K. Yuan and M. Li, Org. Biomol. Chem., 2014, 12, 4628–4632 CAS.
- Z. Chen, H.-Q. Duan, X. Jiang, Y.-M. Zhu, S.-J. Ji and S.-L. Yang, J. Org. Chem., 2015, 80, 8183–8188 CrossRef CAS PubMed.
- L. El Kaim and L. Grimaud, Eur. J. Org. Chem., 2014, 2014, 7749–7762 CrossRef CAS.
- Y. Cheng, X. Yuan, H. Jiang, R. Wang, J. Ma, Y. Zhang and S. Yu, Adv. Synth. Catal., 2014, 356, 2859–2866 CrossRef CAS.
- A. Shaabani, M. Mahyari, M. Aghaei, S. Keshipour and S. W. Ng, Synlett, 2013, 24, 1968–1972 CrossRef CAS.
- Y. Zhang, X. Jiang, J.-M. Wang, J.-L. Chen and Y.-M. Zhu, RSC Adv., 2015, 5, 17060–17063 RSC.
- X. Jiang, J.-M. Wang, Y. Zhang, Z. Chen, Y.-M. Zhu and S.-J. Ji, Tetrahedron, 2015, 71, 4883–4887 CrossRef CAS.
- H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang and S. Yu, Angew. Chem., 2013, 125, 13531–13534 CrossRef.
- E. Soleimani, M. Zainali, N. Ghasemi and B. Notash, Tetrahedron, 2013, 46, 9832–9838 CrossRef.
- X. Wang, S.-Y. Wang and S.-J. Ji, Org. Lett., 2013, 15, 1954–1957 CrossRef CAS PubMed.
- T. Vlaar, P. Mampuys, M. Helliwell, B. U. W. Maes, R. V. A. Orru and E. Ruijter, J. Org. Chem., 2013, 78, 6735–6745 CrossRef CAS PubMed.
- U. K. Sharma, N. Sharma, J. Xu, G. Song and E. V. Van der Eycken, Chem.–Eur. J., 2015, 21, 4908–4912 CrossRef CAS PubMed.
- A. Bazgir and A. M. Astaraki, Eur. J. Chem., 2012, 9, 2315–2321 CAS.
- F. Ji, M.-F. Lv, W.-B. Wen-bin Yi and C. Cai, Synthesis, 2013, 45, 1965–1974 CrossRef CAS.
- A. Shaabani, F. Hajishaabanha, H. Mofakham, M. Mahyari and B. Lali, Helv. Chim. Acta, 2012, 95, 246–254 CrossRef CAS.
- J.-M. Wang, X. Jiang, Y. Zhang, Y.-M. Zhu and J.-K. Shen, Tetrahedron Lett., 2015, 56, 2349–2354 CrossRef CAS.
- S. K. Panja, J. Ghosh, S. Maiti and C. Bandyopadhyay, J. Chem. Res., 2012, 36, 222–225 CrossRef CAS.
- H. Imanieh, M. Sarlak, T. Amanpour and A. Bazgir, Helv. Chim. Acta, 2013, 96, 1978–1982 CrossRef CAS.
- L. Cui, Q. Liu, J. Yu, C. Ni and H. Yu, Tetrahedron Lett., 2011, 52, 5530–5533 CrossRef CAS.
- T. Vlaar, R. C. Cioc, P. Mampuys, B. U. W. Maes, R. V. A. Orru and E. Ruijter, Angew. Chem., 2012, 124, 13235–13238 CrossRef.
- S. Xu, S. Su, H. Zhang, L. Tian, P. Liang, J. Chen, Y. Zhang, C. Li, X. Jia and J. Li, Synthesis, 2015, 47, 2414–2430 CrossRef CAS.
- T.-H. Zhu, S.-Y. Wang, G.-N. Wang and S.-J. Ji, Chem.–Eur. J., 2013, 19, 5850–5853 CrossRef CAS PubMed.
- A. Maleki and A. Sarvary, RSC Adv., 2015, 5, 60938–60955 RSC.
- E. Soleimani, S. Ghorbani and H. R. Ghasempour, Tetrahedron, 2013, 69, 8511–8515 CrossRef CAS.
- L. Shaker Ardakani, M. H. Mosslemin and B. Sadeghi, J. Chem. Res., 2015, 39, 467–469 CrossRef.
- S. Vidyacharan, N. C. Chaitra, A. Sagar and D. S. Sharada, Synth. Commun., 2015, 45, 898–907 CrossRef CAS.
- J. Taran, A. Ramazani, S. W. Joo, K. Slepokura and T. Lis, Helv. Chim. Acta, 2014, 97, 1088–1096 CrossRef CAS.
- I. Yavari, E. Ghanbari and R. Hosseinpour, Helv. Chim. Acta, 2014, 97, 1004–1008 CrossRef CAS.
- S. Sadjadi and M. M. Heravi, Tetrahedron, 2011, 67, 2707–2752 CrossRef CAS.
- V. Jalli, S. Krishnamurthy, H. Kawasaki, T. Moriguchi and A. Tsuge, Synth. Commun., 2015, 45, 2216–2226 CrossRef CAS.
- M. Li, X.-L. Lv, L.-R. Wen and Z.-Q. Hu, Org. Lett., 2013, 15, 1262–1265 CrossRef CAS PubMed.
- G. Qiu, Y. He and J. Wu, Chem. Commun., 2012, 48, 3836–3838 RSC.
- X.-D. Fei, Z.-Y. Ge, T. Tang, Y.-M. Zhu and S.-J. Ji, J. Org. Chem., 2012, 77, 10321–10328 CrossRef CAS PubMed.
- M. Ghandi and N. Zarezadeh, Tetrahedron, 2013, 69, 8668–8674 CrossRef CAS.
- R. S. Borisov, L. G. Voskressensky, A. I. Polyakov, T. N. Borisova and A. V. Varlamov, Synlett, 2014, 25, 0955–0958 CrossRef CAS.
- E. Soleimani and M. Zainali, J. Org. Chem., 2011, 76, 10306–10311 CrossRef CAS PubMed.
- N. Kielland, E. Vicente-Garcia, M. Reves, N. Isambert, M. J. Arevalo and R. Lavilla, Adv. Synth. Catal., 2013, 355, 3273–3284 CrossRef CAS.
- S. S. van Berkel, B. G. M. Bogels, M. A. Wijdeven, B. Westermann and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2012, 2012, 3543–3559 CrossRef CAS.
- N. Shams, A. H. Mosslemin, H. Anaraki-Ardakani and E. Zarenezhad, J. Chem. Res., 2015, 39, 270–273 CrossRef CAS.
- J.-M. Wang, X. Jiang, T. Tang, Y.-M. Zhu and J.-K. Shen, Heterocycles, 2014, 89, 1441–1453 CrossRef CAS.
- S. Rostamnia, RSC Adv., 2015, 5, 97044–97065 RSC.
- M. M. Heravi and S. Moghimi, J. Iran. Chem. Soc., 2011, 8, 306–373 CrossRef CAS.
- R. Ramozzi, N. Chéron, B. Braïda, P. C. Hiberty and P. Fleurat-Lessard, New J. Chem., 2012, 36, 1137–1340 RSC.
- A. Habibi, F. Vafadarnejad and M. A. Armand, J. Heterocycl. Chem., 2013, 50, 887–890 CrossRef CAS.
- B. Neue, R. Reiermann, R. Frohlich, B. Wibbeling, K. Bergander and E.-U. Wurthwein, Eur. J. Org. Chem., 2013, 4944–4952 CrossRef CAS.
- A. A. Esmaeili, S. Shahmansouri, A. Habibi and A. R. Fakhari, Tetrahedron, 2012, 68, 8046–8051 CrossRef CAS.
- S. Dianat, M. Mahdavi, S. Moghimi, A. Mouradzadegun, A. Shafiee and A. Foroumadi, Mol. Diversity, 2015, 19, 797–805 CrossRef CAS PubMed.
- L. El Kaim, L. Grimaud and P. Patil, Org. Lett., 2011, 13, 1261–1263 CrossRef CAS PubMed.
- K. Khandan-Barani, M. T. Maghsoodlou, A. Hassanabadi, M. R. Hosseini-Tabatabaei, J. Saffari and M. Kangani, Res. Chem. Intermed., 2015, 41, 3011–3016 CrossRef CAS.
- M. Tian, Y. He, X. Zhang and X. Fan, J. Org. Chem., 2015, 80, 7447–7455 CrossRef CAS PubMed.
- B. Prasad, S. B. Nallapati, S. K. Kolli, A. K. Sharma, S. Yellanki, R. Medisetti, P. Kulkarni, S. Sripelly, K. Mukkanti and M. Pal, RSC Adv., 2015, 5, 62966–62970 RSC.
- T.-H. Zhu, X. Zhu, X.-P. Xu, T. Chen and S.-J. Ji, Tetrahedron Lett., 2011, 52, 2771–2775 CrossRef CAS.
- I. Yavari, R. Pashazadeh and R. Hosseinpour, Helv. Chim. Acta, 2012, 95, 169–172 CrossRef CAS.
- R. Wang, X.-P. Xu, H. Meng, S.-Y. Wang and S.-J. Ji, Tetrahedron, 2013, 69, 1761–1766 CrossRef CAS.
- K. Tamura, N. Kumagai and M. Shibasaki, Eur. J. Org. Chem., 2015, 2015, 3026–3031 CrossRef CAS.
- X. Qi, H. Zhang, A. Shao, L. Zhu, T. Xu, M. Gao, C. Liu and Y. Lan, ACS Catal., 2015, 5, 6640–6647 CrossRef CAS.
- P. Xiao, H. Yuan, J. Liu, Y. Zheng, X. Bi and J.-P. Zhang, ACS Catal., 2015, 5, 6177–6184 CrossRef CAS.
- T. Buyck, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2014, 136, 11524–11528 CrossRef CAS PubMed.
- A. Coppola, D. Sucunza, C. Burgos and J. J. Vaquero, Org. Lett., 2015, 17, 78–81 CrossRef CAS PubMed.
- P. C. Knipe, M. Gredicak, A. Cernijenko, R. S. Paton and M. D. Smith, Chem.–Eur. J., 2014, 20, 3005–3009 CrossRef CAS PubMed.
- L. Hu, W. Gui, Z. Liu and B. Jiang, RSC Adv., 2014, 4, 38258–38262 RSC.
- S. Su, C. Li, X. Jia and J. Li, Chem.–Eur. J., 2014, 20, 5905–5909 CrossRef CAS PubMed.
- X. Meng, P. Liao, J. Liu and X. Bi, Chem. Commun., 2014, 50, 11837–11839 RSC.
- V. Estévez, G. Van Baelen, B. H. Lentferink, T. Vlaar, E. Janssen, B. U. W. Maes, R. V. A. Orru and E. Ruijter, ACS Catal., 2014, 4, 40–43 CrossRef.
- G. Koopmanschap, E. Ruijter and R. V. A. Orru, Beilstein J. Org. Chem., 2014, 10, 544–598 CrossRef PubMed.
- A. M. Astaraki and A. Bazgir, J. Heterocycl. Chem., 2013, 50, 175–178 CrossRef CAS.
- S. Kim and S. H. Hong, Adv. Synth. Catal., 2015, 357, 1004–1012 CrossRef CAS.
- F. Sha, L. Wu and X. Huang, J. Org. Chem., 2012, 77, 3754–3765 CrossRef CAS PubMed.
- K. Perez-Labrada, I. Brouard, I. Mendez and D. G. Rivera, J. Org. Chem., 2012, 77, 4660–4670 CrossRef CAS PubMed.
- S. Zhang, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2013, 52, 3485–3489 CrossRef CAS PubMed.
- A. Zajdlik, Z. Wang, J. L. Hickey, A. Aman, A. D. Schimmer and A. K. Yudin, Angew. Chem., Int. Ed., 2013, 52, 8411–8415 CrossRef CAS PubMed.
- A. F. Maddox, J. J. Davidson, T. Shalumova, J. M. Tanski and R. Waterman, Inorg. Chem., 2013, 52, 7811–7816 CrossRef CAS PubMed.
- G. Qiu, X. Qiu and J. Wua, Adv. Synth. Catal., 2013, 355, 3205–3209 CrossRef CAS.
- T. Mitamura, K. Iwata and A. Ogawa, J. Org. Chem., 2011, 76, 3880–3887 CrossRef CAS PubMed.
- J. Li, Y. He, S. Luo, J. Lei, J. Wang, Z. Xie and Q. Zhu, J. Org. Chem., 2015, 80, 2223–2230 CrossRef CAS PubMed.
- Y. Ohmori, M. Ichinohe, A. Sekiguchi, M. J. Cowley, V. Huch and D. Scheschkewitz, Organometallics, 2013, 32, 1591–1594 CrossRef CAS.
- S. Wiese, M. J. B. Aguila, E. Kogut and T. H. Warren, Organometallics, 2013, 32, 2300–2308 CrossRef CAS.
- E. R. Klobukowski, R. J. Angelici and L. K. Woo, Organometallics, 2012, 31, 2785–2792 CrossRef CAS.
- R. Akbarzadeh, T. Amanpour, H. R. Khavasi and A. Bazgir, Tetrahedron, 2014, 70, 169–175 CrossRef CAS.
- L. Zhang, X. Xu, Q.-R. Shao, L. Pana and Q. Liu, Org. Biomol. Chem., 2013, 7393–7399 CAS.
- L. El Kaim, L. Grimaud and P. Pravin, Org. Lett., 2012, 14, 476–478 CrossRef CAS PubMed.
- M. K. Sinha, K. Khoury, E. Herdtweck and A. Domling, Chem.–Eur. J., 2013, 19, 8048–8052 CrossRef CAS PubMed.
- M. K. Sinha, K. Khoury, E. Herdtweck and A. Domling, Org. Biomol. Chem., 2013, 11, 4792–4796 CAS.
- A. G. Neo, J. Díaz, S. Marcaccini and C. F. Marcos, Org. Biomol. Chem., 2012, 10, 3406–3416 CAS.
- L. Zhang, X. Xu, W. Xia and Q. Liu, Adv. Synth. Catal., 2011, 353, 2619–2623 CrossRef CAS.
- M.-J. Oliva-Madrid, J.-A. Garcıá -Lopez, I. Saura-Llamas, D. Bautista and J. Vicente, Organometallics, 2012, 31, 3647–3660 CrossRef CAS.
- R. Frutos-Pedreno, P. Gonzalez-Herrero and J. Vicente, Organometallics, 2013, 32, 4664–4676 CrossRef CAS.
- A. Abellan-Lopez, M.-T. Chicote, D. Bautista and J. Vicente, Organometallics, 2013, 32, 7612–7624 CrossRef CAS.
- R. Frutos-Pedreño, P. González-Herrero and J. Vicente, Organometallics, 2012, 31, 3361–3372 CrossRef.
- J. Vicente, J. A. Abad and R.-M. Lopez-Nicolas, Organometallics, 2011, 30, 4983–4998 CrossRef CAS.
- M. M. Heravi and T. Alishiri, Adv. Heterocycl. Chem., 2014, 113, 1–66 CrossRef CAS.
- M. M. Heravi and B. Talaei, Adv. Heterocycl. Chem., 2014, 113, 143–244 CrossRef CAS.
- M. M. Heravi, S. Khaghaninejad and M. Mostofi, Adv. Heterocycl. Chem., 2014, 112, 1–50 CrossRef CAS.
- M. M. Heravi, S. Khaghaninejad and N. Nazari, Adv. Heterocycl. Chem., 2014, 112, 183–234 CrossRef CAS.
- S. Khaghaninejad and M. M. Heravi, Adv. Heterocycl. Chem., 2014, 111, 95–146 CrossRef CAS.
- M. M. Heravi and V. Zadsirjan, Adv. Heterocycl. Chem., 2015, 117, 261–376 CrossRef.
- M. M. Heravi and B. Talaei, Adv. Heterocycl. Chem., 2015, 114, 147–225 CrossRef.
- M. M. Heravi and V. Fathi Vavsari, Adv. Heterocycl. Chem., 2015, 114, 77–145 CrossRef.
- M. M. Heravi, S. Asadi and B. M. Lashkariani, Mol. Diversity, 2013, 17, 389–407 CrossRef CAS PubMed.
- M. M. Heravi, F. Derikvand and F. F. Bamoharram, J. Mol. Catal. A: Chem., 2007, 263, 112–114 CrossRef CAS.
- M. M. Heravi, B. Baghernejad and H. A. Oskooie, Mol. Diversity, 2009, 13, 395–398 CrossRef CAS PubMed.
- M. M. Heravi, B. Baghernejad, H. A. Oskooie and R. Hekmatshoar, Tetrahedron Lett., 2008, 6101–6103 CrossRef CAS.
- M. M. Heravi, B. Baghernejad and H. A. oskooie, Synlett, 2009, 1123–1125 CrossRef CAS.
- G. Mohammadi Ziarani, Z. Dashtianeh, M. Shakiba Nahad and A. Badiei, Arabian J. Chem., 2015, 8, 692–697 CrossRef CAS.
- M. M. Heravi, L. Ranhbar, F. Derikvand and F. F. Bamoharram, J. Mol. Catal. A: Chem., 2007, 276, 226–229 CrossRef CAS.
- F. Nemati, M. M. Heravi and A. Elhampour, RSC Adv., 2015, 5, 45775–45784 RSC.
- M. M. Heravi, F. Mousavizadeh, N. Ghobadi and M. Tajbakhsh, Tetrahedron Lett., 2014, 55, 1226–1228 CrossRef CAS.
- M. M. Heravi, E. Hashemi, Y. S. Beheshtiha, K. Kamjou, M. Toolabi and N. Hosseintash, J. Mol. Catal. A: Chem., 2014, 392, 173–180 CrossRef CAS.
- M. M. Heravi, B. Baghernejad and H. A. Oskooie, Tetrahedron Lett., 2009, 50, 767–769 CrossRef CAS.
- M. M. Heravi, F. Derikvand and M. Haghighi, Monatsh. Chem., 2007, 139, 31–33 CrossRef.
- M. M. Heravi and E. Hashemi, Tetrahedron, 2012, 68, 9145–9178 CrossRef CAS.
- M. M. Heravi, E. Hashemi and F. Azimian, Tetrahedron, 2014, 70, 7–21 CrossRef CAS.
- M. M. Heravi and V. Zadsirjan, Tetrahedron: Asymmetry, 2013, 24, 1149–1188 CrossRef CAS.
- M. M. Heravi, E. Hashemi and N. Ghobadi, Curr. Org. Chem., 2013, 17, 2192–2224 CrossRef CAS.
- M. M. Heravi, E. Hashemi and N. Nazari, Mol. Diversity, 2014, 18, 441–472 CrossRef CAS PubMed.
- M. M. Heravi, V. Zadsirjan and Z. Bozorgpour Savadjani, Curr. Org. Chem., 2014, 18, 2857–2891 CrossRef CAS.
- M. M. Heravi and V. Fathi Vavsari, RSC Adv., 2015, 5, 50890–50912 RSC.
- M. M. Heravi, A. Bakhtiari and Z. Faghihi, Curr. Org. Synth., 2014, 11, 787–823 CrossRef CAS.
- M. M. Heravi, T. Baie Lashaki and N. Poorahmad, Tetrahedron: Asymmetry, 2015, 26, 405–495 CrossRef CAS.
- M. M. Heravi, M. V. Fard and Z. Faghihi, Curr. Org. Chem., 2015, 19, 1491–1525 CrossRef CAS.
- M. M. Heravi and A. Fazeli, Heterocycles, 2010, 81, 1979–2026 CrossRef CAS.
- M. M. Heravi, M. Vazin Fard and Z. Faghihi, Green Chem. Lett. Rev., 2013, 6, 282–300 CrossRef CAS.
- M. M. Heravi and P. Hajiabbasi, Monatsh. Chem., 2012, 143, 1575–1592 CrossRef CAS.
- M. M. Heravi, B. Baghernejad and H. A. Oskooie, Curr. Org. Chem., 2009, 13, 1002–1014 CrossRef CAS.
- M. M. Heravi and S. Sadjadi, J. Iran. Chem. Soc., 2009, 6, 1–54 CrossRef CAS.
- M. M. Heravi, T. Ahmadi, A. Fazeli and N. M. Kalkhorani, Curr. Org. Synth., 2015, 12, 328–357 CrossRef CAS.
- M. M. Heravi, A. Fazeli and Z. Faghihi, Curr. Org. Synth., 2016, 13, 220–254 CrossRef CAS.
- M. M. Heravi, T. Ahmadi, A. Fazeli and N. M. Kalkhorani, Curr. Org. Synth., 2015, 12, 328–357 CrossRef CAS.
- H. Braunschweig, T. Herbst, K. Radacki, C. W. Tate and A. Vargas, Chem. Commun., 2013, 49, 1702–1704 RSC.
- T. Soeta, Y. Miyamoto, S. Fujinami and Y. Ukaji, Tetrahedron, 2014, 70, 6623–6629 CrossRef CAS.
- J. Liu, Z. Fang, Q. Zhang, Q. Liu and X. Xihe Bi, Angew. Chem., Int. Ed., 2013, 52, 6953–6957 CrossRef CAS PubMed.
- X. Wang, X.-P. Xu, S.-Y. Wang, W. Zhou and S.-J. Ji, Org. Lett., 2013, 15, 4246–4249 CrossRef CAS PubMed.
- M. Adib, B. Mohammadi, E. Sheikhi and H. R. Bijanzadeh, Chin. Chem. Lett., 2011, 22, 314–317 CrossRef CAS.
- F. Qiu, J. Wu, Y. Zhang, M. Hub and Y. Yu, Tetrahedron Lett., 2012, 53, 446–448 CrossRef CAS.
- F. Qiu, J. Wu, Y. Zhang, M. Hu and Y. Yu, Tetrahedron Lett., 2012, 53, 446–448 CrossRef CAS.
- I. Yavari and M. Nematpour, Helv. Chim. Acta, 2013, 96, 2098–2102 CrossRef CAS.
- I. Yavari, R. Hosseinpour and R. Pashazadeh, Synlett, 2012, 23, 1662–1666 CrossRef CAS.
- W. Chen, J. Shao, Z. Li, M. A. Giulianotti and Y. Yu, Can. J. Chem., 2012, 90, 214–221 CrossRef CAS.
- M. Gao, C. He, H. Chen, R. Bai, B. Cheng and A. Lei, Angew. Chem., Int. Ed., 2013, 125, 6958–6961 CrossRef PubMed.
- J. Liu, Z. Fang, Q. Zhang, Q. Liu and X. Bi, Angew. Chem., Int. Ed., 2013, 52, 6953–6957 CrossRef CAS PubMed.
- F. Qiu, J. Wu, Y. Zhang, M. Hu and Y. Yu, Tetrahedron Lett., 2012, 53, 446–448 CrossRef CAS.
- X. Xin, X. Liu, D. Zhang, R. Zhang, Y. Liang, F. Han and D. Dong, Org. Biomol. Chem., 2014, 12, 5477–5483 CAS.
- M. Adib, B. Mohammadi, E. Sheikhi and H. R. Bijanzadeh, Chin. Chem. Lett., 2011, 22, 314–317 CrossRef CAS.
- S. Yugandar, N. C. Misra, G. Parameshwarappa, K. Panda and H. Ila, Org. Lett., 2013, 15, 5250–5253 CrossRef CAS PubMed.
- F. Zhou, J. Liu, K. Ding, J. Liu and Q. Cai, J. Org. Chem., 2011, 76, 5346–5353 CrossRef CAS PubMed.
- A. Znabet, S. Blanken, E. Janssen, F. J. J. de Kanter, M. Helliwell, N. J. Turner, E. Ruijter and R. V. A. Orru, Org. Biomol. Chem., 2012, 10, 941–944 CAS.
- Y. Li, J. Zhao, H. Chen, B. Liu and H. Jiang, Chem. Commun., 2012, 48, 3545–3547 RSC.
- J.-Y. Liao, P.-L. Shao and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 628–631 CrossRef CAS PubMed.
- S. Jia, S. Su, C. Li, X. Jia and J. Li, Org. Lett., 2014, 16, 5604–5607 CrossRef CAS PubMed.
- J. Peng, J. Zhao, Z. Hu, D. Liang, J. Huang and Q. Zhu, Org. Lett., 2012, 14, 4966–4969 CrossRef CAS PubMed.
- J. Peng, L. Liu, Z. Hu, J. Huang and Q. Zhu, Chem. Commun., 2012, 48, 3772–3774 RSC.
- Y.-Y. Pan, Y.-N. Wu, Z.-Z. Chen, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, J. Org. Chem., 2015, 80, 5764–5770 CrossRef CAS PubMed.
- J. Liu, Z. Liu, P. Liao and X. Bi, Org. Lett., 2014, 16, 6204–6207 CrossRef CAS PubMed.
- H. Jie, J. Li, C. Li and X. Jia, Synlett, 2012, 23, 2274–2278 CrossRef CAS.
- J. Li, S. Su, M. Huang, B. Song, C. Li and X. Jia, Chem. Commun., 2013, 49, 10694–10696 RSC.
- H. Jia, J. Li, C. Li and X. Jia, Synlett, 2012, 23, 2274–2278 CrossRef.
- G. Qiu, X. Qiu, J. Liu and J. Wu, Adv. Synth. Catal., 2013, 355, 2441–2446 CrossRef CAS.
- Y. Li, X. Xu, C. Xia, L. Zhang, L. Pan and Q. Liu, Chem. Commun., 2012, 48, 12228–12230 RSC.
- T. Nanjo, C. Tsukano and Y. Takemoto, Org. Lett., 2012, 14, 4270–4273 CrossRef CAS PubMed.
- T. Nanjo, C. Tsukano and Y. Takemoto, Synlett, 2014, 25, 1473–1477 CrossRef.
- T. Nanjo, S. Yamamoto, C. Tsukano and Y. Takemoto, Org. Lett., 2013, 15, 3754–3757 CrossRef CAS PubMed.
- G. Qiu, X. Qiu, J. Liu and J. Wu, Adv. Synth. Catal., 2013, 355, 2441–2446 CrossRef CAS.
- Y. Wang and Q. Qiang Zhu, Adv. Synth. Catal., 2012, 354, 1902–1908 CrossRef CAS.
- Z. Hu, J. Wang, D. Liang and Q. Zhu, Adv. Synth. Catal., 2013, 355, 3290–3294 CrossRef CAS.
- T. Tang, X. Jiang, J.-M. Wang, Y.-X. Sun and Y.-M. Zhu, Tetrahedron, 2014, 70, 2999–3004 CrossRef CAS.
- G. Kaur, A. Vadekeetil, K. Harjai and V. Singh, Tetrahedron Lett., 2015, 56, 4445–4450 CrossRef CAS.
- H. Wang, Y.-L. Zhao, C.-Q. Ren, A. Diallo and Q. Liu, Chem. Commun., 2011, 47, 12316–12318 RSC.
- G. Qiu, C. Chen, L. Yao and J. Wu, Adv. Synth. Catal., 2013, 355, 1579–1584 CrossRef CAS.
- T. Miura, Y. Nishida, M. Morimoto, M. Yamauchi and M. Murakami, Org. Lett., 2011, 13, 1429–1431 CrossRef CAS PubMed.
- B. Liu, Y. Li, H. Jiang, M. Yin and H. Huang, Adv. Synth. Catal., 2012, 354, 2288–2300 CrossRef CAS.
- Q. Dai, Y. Jiang, S. Guo, J.-T. Yu and J. Cheng, Chem. Commun., 2015, 51, 14781–14784 RSC.
- Q. Gao, P. Zhou, F. Liu, W.-J. Hao, C. Yao, B. Jiang and S.-J. Tu, Chem. Commun., 2015, 51, 9519–9522 RSC.
- M. Shiri, S. Z. Mirpour-Marzoni, Z. Bozorgpour-Savadjani, B. Soleymanifard and H. G. Kruger, Monatsh. Chem., 2014, 145, 1947–1952 CrossRef CAS.
- S. J. Welsch, M. Umkehrer, G. Ross, J. Kolb, C. Burdack and L. A. Wessjohann, Tetrahedron Lett., 2011, 52, 6295–6297 CrossRef CAS.
- A. Sarvary, S. Shaabani, A. Shaabani and S. W. Ng, Tetrahedron, 2011, 67, 3624–3630 CrossRef CAS.
- A. Shaabani, M. Mahyari, F. Hajishaabanha and H. Mofakham, J. Iran. Chem. Soc., 2014, 11, 1183–1187 CrossRef CAS.
- F. Gharkhani, E. Vessally, A. Mohammadi and Z. Alimadadi, Asian J. Chem., 2013, 25, 7647–7648 Search PubMed.
- A. Habibi and A. Rahmani, Helv. Chim. Acta, 2011, 94, 1806–1811 CrossRef CAS.
- S. Asghari, Z. Tayebi and A. K. Habibi, J. Heterocycl. Chem., 2013, 50, 874–878 CrossRef CAS.
- M. Kumar, L. Kumar Kumawat, V. Kumar Gupta and A. Sharma, ChemistryOpen, 2015, 4, 626–632 CrossRef CAS PubMed.
- K. Khandan–Barani, M. T. Maghsoodlou, S. M. Habibi-Khorasani, N. Hazeri and S. S. Sajadikhah, J. Chem. Res., 2011, 35, 231–233 CrossRef.
- S. Damavandi, R. Sandaroos and M. Pashirzad, Res. Chem. Intermed., 2012, 38, 1969–1974 CrossRef CAS.
- A. A. Esmaeili, M. Zangouei, R. Hosseinabadi and A. R. Fakhari, Mol. Diversity, 2012, 16, 145–150 CrossRef CAS PubMed.
- W. Han, J. Wu and W.-M. Dai, Synlett, 2014, 25, 2019–2024 CrossRef CAS.
- S. Keshipour, S. Shaabani and A. Shaabani, Tetrahedron Lett., 2012, 53, 7085–7087 CrossRef CAS.
- F. Matloubi Moghaddam, M. R. Khodabakhshi and A. Latifkar, Tetrahedron Lett., 2014, 55, 1251–1254 CrossRef CAS.
- O. Ghashghaei, M. Revés, N. Kielland and R. Lavilla, Eur. J. Org. Chem., 2015, 4383–4388 CrossRef CAS.
- R. Bujok, P. Cmoch and Z. Wróbel, Tetrahedron Lett., 2014, 55, 3410–3413 CrossRef CAS.
- A. S. Bunev, M. A. Vasiliev, V. E. Statsyuk, G. I. Ostapenko and A. S. Peregudov, J. Fluorine Chem., 2014, 163, 34–37 CrossRef CAS.
- S. Rostamnia and A. Hassankhani, RSC Adv., 2013, 3, 18626–18629 RSC.
- A. Demjen, M. Gyuris, J. Wolfling, L. G. Puskas and I. Kanizsai, Beilstein J. Org. Chem., 2014, 10, 2338–2344 CrossRef PubMed.
- M. Anary-Abbasinejad, N. Shams and M. Heidari, ARKIVOC, 2012, 13–20 CAS.
- T.-H. Zhu, T.-Q. Wei, S.-Y. Wang and S.-J. Ji, Org. Chem. Front., 2015, 2, 259–264 RSC.
- S. Yugandar, A. Acharya and H. Ila, J. Org. Chem., 2013, 78, 3948–3960 CrossRef CAS PubMed.
- L. El Kaim, L. Grimaud and P. Patil, Synlett, 2012, 23, 1361–1363 CrossRef CAS.
- I. Yavari, T. Sanaeishoar, L. Azad and M. Ghazvini, Mendeleev Commun., 2011, 21, 108–109 CrossRef CAS.
- I. Yavari, R. Pashazadeh, R. Hosseinpour and E. Ghanbari, Tetrahedron Lett., 2013, 54, 2785–2787 CrossRef CAS.
- M. Mahdavi, M. Asadi, M. Saeedi, M. Ebrahimi, M. A. Rasouli, P. R. Ranjbar, A. Foroumadi and A. Shafiee, Synthesis, 2012, 44, 3649–3654 CrossRef CAS.
- I. Yavari, S. Arab-Salmanabadi and A. Aminkhani, Chin. Chem. Lett., 2012, 23, 49–52 CrossRef CAS.
- T.-H. Zhu, X. Zhu, X.-P. Xu, T. Chen and S.-J. Ji, Tetrahedron Lett., 2011, 52, 2771–2775 CrossRef CAS.
- I. Yavari a, R. Hosseinpour, R. Pashazadeh, E. Ghanbari and S. Skoulika, Tetrahedron, 2013, 69, 2462–2467 CrossRef.
- P. Dang, W. Zeng and Y. Liang, Org. Lett., 2015, 17, 34–37 CrossRef CAS PubMed.
- X.-Y. Fan, X. Jiang, Y. Zhang, Z.-B. Chen and Y.-M. Zhu, Org. Biomol. Chem., 2015, 13, 10402–10408 CAS.
- M. Rouhani, A. Ramazani and W. Joo, Ultrason. Sonochem., 2015, 22, 391–396 CrossRef CAS PubMed.
- H. Yanai, T. Sakiyama, T. Oguchi and T. Taguchi, Tetrahedron Lett., 2012, 53, 3161–3164 CrossRef CAS.
- T. Amanpour, P. Mirzaei and A. Bazgir, Tetrahedron Lett., 2012, 53, 1421–1423 CrossRef CAS.
- A. R. Kazemizadeh, N. Hajaliakbari, R. Hajian, N. Shajari and A. Ramazani, Helv. Chim. Acta, 2012, 95, 594–597 CrossRef CAS.
- C.-H. Lei, D.-X. Wang, L. Zhao, J. Zhu and M.-X. Wang, Chem.–Eur. J., 2013, 19, 16981–16987 CrossRef CAS PubMed.
- F. Sha, H. Shen and X.-Y. Wu, Eur. J. Org. Chem., 2013, 2537–2540 CrossRef CAS.
- M. Adib, E. Sheikhi and N. Rezaei, Tetrahedron Lett., 2011, 52, 3191–3194 CrossRef CAS.
- A. B. Ramesha, G. M. Raghavendra, K. N. Nandeesh, K. S. Rangappa and K. Mantelingu, Tetrahedron Lett., 2013, 54, 95–100 CrossRef CAS.
- S. K. Guchhait, G. Priyadarshani, V. Chaudhary, D. R. Seladiya, T. M. Shah and N. P. Bhogayta, RSC Adv., 2013, 3, 10867–10874 RSC.
- B. Mohtat, H. Djahaniani, R. Khorrami, S. Mashayekhi and I. Yavari, Synth. Commun., 2011, 41, 784–791 CrossRef CAS.
- A. T. Khan, R. S. Basha and M. Lal, Tetrahedron Lett., 2012, 53, 2211–2217 CrossRef CAS.
- M. Li, W. Kong, L.-R. Wen and F.-H. Liu, Tetrahedron, 2012, 68, 4838–4845 CrossRef CAS.
- B. Liu, H. Gao, Y. Yu, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 10319–10328 CrossRef CAS PubMed.
- Q. Zheng, Q. Ding, X. Liu, Y. Zhang and Y. Peng, J. Organomet. Chem., 2015, 783, 77–82 CrossRef CAS.
- Z.-Y. Gu, T.-H. Zhu, J.-J. Cao, X.-P. Xu, S.-Y. Wang and S.-J. Ji, ACS Catal., 2014, 4, 49–52 CrossRef CAS.
- Z. Hu, Y. Li, L. Pan and X. Xu, Adv. Synth. Catal., 2014, 356, 2974–2978 CrossRef CAS.
- T. Mitamura and A. Ogawa, J. Org. Chem., 2011, 76, 1163–1166 CrossRef CAS PubMed.
- T. Soeta, S. Fujinami and Y. Ukaji, J. Org. Chem., 2012, 77, 9878–9883 CrossRef CAS PubMed.
- M. A. Rasouli, M. Mahdavi, M. Saeedi, P. Rashidi Ranjbar, A. Shafiee and A. Foroumadi, J. Heterocycl. Chem., 2015, 52, 386–391 CrossRef CAS.
- M. A. Rasouli, M. Mahdavi, L. Firoozpour, A. Shafiee and A. Foroumadi, Tetrahedron, 2014, 70, 3931–3934 CrossRef CAS.
- T. Wang, R. Li, D. Yu, C. Gu, F. Xiong and Z. Chen, Synthesis, 2014, 46, 3213–3220 CrossRef CAS.
- R. Wang, S.-Y. Wang and S.-J. Ji, Tetrahedron, 2013, 69, 10836–10841 CrossRef CAS.
- V. Kavala, C.-C. Wang, Y.-H. Wang, C.-W. Kuo, D. Janreddy, W.-C. Huang, T.-S. Kuo, C. H. He, M.-L. Chen and C.-F. Yao, Adv. Synth. Catal., 2014, 356, 2609–2626 CrossRef CAS.
- T. Soeta, K. Tamura, S. Fujinami and Y. Ukaji, Org. Biomol. Chem., 2013, 11, 2168–2174 CAS.
- A. Aminkhani, Heterocycl. Commun., 2013, 19, 109–112 CrossRef CAS.
- H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2013, 52, 13289–13292 CrossRef CAS PubMed.
- J. Azizian, A. Ramazani and M. Haji, Helv. Chim. Acta, 2011, 94, 371–375 CrossRef CAS.
- A. Hassanabadi, M. H. Mosslemin, M. Anary-Abbasinejad and M. Ghasemi, Synth. Commun., 2011, 41, 3714–3719 CrossRef CAS.
- B. Mohtat, H. Djahaniani, I. Yavari, M. G. Dehbalaei and S. A. Jam, Chin. Chem. Lett., 2011, 22, 771–773 CrossRef CAS.
- M. B. Teimouri, P. Akbari-Moghaddam and G. Golbaghi, ACS Comb. Sci., 2011, 13, 659–666 CrossRef CAS PubMed.
- M. B. Teimouri, Tetrahedron, 2011, 67, 1837–1843 CrossRef CAS.
- M. Gyuris, R. Madacsi, L. G. Puskás, G. K. Toth, J. Wolfling and I. Kanizsai, Eur. J. Org. Chem., 2011, 848–851 CrossRef CAS.
- S. Keshipour, A. Shaabani, S. Shojaei, H. Nosrati and S. W. Ng, J. Iran. Chem. Soc., 2015, 12, 1655–1663 CrossRef CAS.
- A. G. Neo, T. G. Castellano and C. F. Marcos, Synthesis, 2015, 26, 2431–2438 CrossRef.
- A. Hossein Rezayan, C. R. Chim., 2012, 15, 499–503 CrossRef.
- M. Giustiniano, V. Mercalli, J. Amato, E. Novellino and G. C. Tron, Org. Lett., 2015, 17, 3964–3967 CrossRef CAS PubMed.
- S. Keshipour, S. Shojaei and A. Shaabani, Tetrahedron, 2012, 68, 6141–6145 CrossRef CAS.
- M. T. Maghsoodlou, G. Marandi, N. Hazeri, S. M. Habibi-Khorassani and A. A. Mirzaei, Mol. Diversity, 2011, 15, 227–231 CrossRef CAS PubMed.
- A. Habibi and Z. Tarameshloo, J. Iran. Chem. Soc., 2011, 8, 287–291 CrossRef CAS.
- D. Zheng and J. Wu, Eur. J. Org. Chem., 2014, 767–771 CrossRef CAS.
- A. A. Esmaeili, M. Zangouei, A. R. Fakhari and A. Habibi, Tetrahedron Lett., 2012, 53, 1351–1353 CrossRef CAS.
- M. Zangouei, A. A. Esmaeili, A. Habibi and A. R. Fakhari, Tetrahedron, 2014, 70, 8619–8623 CrossRef CAS.
- S. Sharma and A. Jain, Tetrahedron Lett., 2014, 55, 6051–6054 CrossRef CAS.
- G. Qiu, G. Liu, S. Pu and J. Wu, Chem. Commun., 2012, 48, 2903–2905 RSC.
- T. Zarganes-Tzitzikas, P. Patil, K. Khoury, E. Herdtweck and A. Domling, Eur. J. Org. Chem., 2015, 51–55 CrossRef CAS PubMed.
- M. Shiri and Z. Bozorgpour-Savadjani, J. Iran. Chem. Soc., 2015, 12, 389–396 CrossRef CAS.
- F. Ji, W.-B. Yi and C. Cai, J. Heterocycl. Chem., 2014, 51, 1287–1292 CrossRef CAS.
- T. Amanpour, P. Mirzaei and A. Bazgir, Synthesis, 2012, 44, 235–240 CAS.
- R. Akbarzadeh, T. Amanpour, P. Mirzaei and A. Bazgir, Helv. Chim. Acta, 2011, 94, 1527–1532 CrossRef CAS.
- T. Vlaar, R. V. A. Orru, B. W. Maes and E. Ruijter, J. Org. Chem., 2013, 78, 10469–10475 CrossRef CAS PubMed.
- R. Baharfar and S. M. Baghbanian, J. Heterocycl. Chem., 2012, 49, 310–314 CrossRef CAS.
- B. Liu, M. Yin, H. Gao, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3009–3020 CrossRef CAS PubMed.
- W. Hao, J. Zeng and M. Cai, Chem. Commun., 2014, 50, 11686–11689 RSC.
- R. Baharfar, S. M. Baghbanian and S. M. Vahdat, Tetrahedron Lett., 2011, 52, 6018–6020 CrossRef CAS.
- G. Pandey, S. Bhowmik and S. Batra, RSC Adv., 2014, 4, 41433–41436 RSC.
- T. Soeta, K. Tamura and Y. Ukaji, Org. Lett., 2012, 14, 1226–1229 CrossRef CAS PubMed.
- R. Akbarzadeh, T. Amanpour, A. Abolhasani Soorki and A. Bazgir, Helv. Chim. Acta, 2012, 95, 483–490 CrossRef CAS.
- D. Bhattacharya, S. Mitra and P. Chattopadhyay, Synthesis, 2015, 47, 2294–2298 CrossRef CAS.
- B. Liu, Y. Li, M. Yin, W. Wu and H. Jiang, Chem. Commun., 2012, 48, 11446–11448 RSC.
- M. Ghandi, N. Zarezadeh and A. Taheri, Tetrahedron Lett., 2011, 52, 1228–1232 CrossRef CAS.
- G. Kulsi, A. Ghorai and P. Chattopadhyay, Tetrahedron Lett., 2012, 53, 3619–3622 CrossRef CAS.
- B. H. Rotstein, D. J. Winternheimer, L. M. Yin, C. M. Deber and A. K. udin, Chem. Commun., 2012, 48, 3775–3777 RSC.
- M. Bachman, S. E. Mann and T. D. Sheppard, Org. Biomol. Chem., 2012, 10, 162–170 CAS.
- F. De Moliner, M. Bigatti, C. De Rosa, L. Banfi, R. Riva and A. Basso, Mol. Diversity, 2014, 18, 473–482 CrossRef CAS PubMed.
- D. Yugandhar and A. Kumar Srivastava, ACS Comb. Sci., 2015, 17, 474–481 CrossRef CAS PubMed.
- H. R. Reza Safaei, N. Shioukhi and M. Shekouhy, Monatsh. Chem., 2013, 144, 1855–1863 CrossRef.
- E. Soleimani, M. Zainali, N. Ghasemi and B. Notash, Tetrahedron, 2013, 69, 9832–9838 CrossRef CAS.
- R. Baharfar, L. Jaafari and R. Azimi, Chin. Chem. Lett., 2011, 22, 943–946 CrossRef CAS.
- A. Bornadiego, J. Diaz and C. F. Marcos, Adv. Synth. Catal., 2014, 356, 718–722 CrossRef CAS.
- J. Ruiz, M. P. Gonzalo, M. Vivanco, M. R. Diaz and S. Garcia-Granda, Chem. Commun., 2011, 47, 4270–4272 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.