
Nanoparticle supported, magnetically separable manganese porphyrin as an efficient retrievable nanocatalyst in hydrocarbon oxidation reactions†
Mojtaba Bagherzadeh* and
Anahita Mortazavi-Manesh
Chemistry Department, Sharif University of Technology, P. O. Box 11155-3615, Tehran, Iran. E-mail: bagherzadeh@sharif.edu; Fax: +98 21 66012983; Tel: +98 21 66165354
First published on 14th April 2016
Abstract
A manganese porphyrin, meso-tetrakis(pentafluorophenyl)porphyrinato manganese(III) acetate, Mn(TPFPP)OAc, was immobilized on silica-coated magnetic Fe3O4 nanoparticles functionalized with 3-aminopropyltriethoxysilane (APTS) through the amino propyl linkage using a grafting process in toluene solvent. This enabled the covalent immobilization of Mn(III) porphyrin via an aromatic nucleophilic substitution reaction, to afford the Fe3O4@SiO2–NH2@MnPor catalyst. The resulting nanoparticles were characterized by X-ray powder diffraction (XRD), scanning electron microscopy (SEM), FT-IR spectroscopy, UV-Vis spectroscopy, elemental analysis (CHN), atomic absorption spectroscopy (AAS), and vibrating sample magnetometry (VSM). The immobilized manganese porphyrin was applied as an efficient and retrievable heterogeneous nanocatalyst in alkane hydroxylation and alkene epoxidation. Leaching and recycling experiments revealed that the prepared nanocatalyst can be recovered, and reused several times, without loss of activity and magnetic properties.
1. Introduction
Metalloporphyrins are well known to mimic the activity of enzymatic monooxygenases.1,2 In this regard, metalloporphyrin complexes have been largely employed as valuable biomimetic catalysts, owing to the critical roles they play in oxygen transfer processes.3 Investigating in this area is based on different strategies to design selective, stable and high turnover catalytic systems.4,5 The introduction of second-generation metalloporphyrins, which bear bulky and/or electron-withdrawing substituents at the meso positions of the porphyrin ring, has been among the main approaches commonly employed with the aim of increasing the stability of the aromatic heterocycle towards oxidative degradation in reaction conditions. Immobilization of expensive metalloporphyrin catalysts onto supports appears to be a good way to improve their oxidative stability, selectivity and the catalytic performance because of the support environment and other advantages with respect to recovery and reuse.6–11 In other words, supporting metalloporphyrins provides a physical separation of active sites, thus minimizing catalyst self-destruction and metalloporphyrin dimerization.10,12 Furthermore, heterogeneous catalytic oxidations have become an important target since their process are used in industry, helping to minimize the problems of industrial waste treatment. Hence, the immobilization of these biomimetic catalysts is much desired.An attractive approach is the preparation of magnetically separable nanocatalysts.13–16 In this manner, silica-coated magnetic nanoparticles, Fe3O4@SiO2, have been studied extensively due to their superparamagnetism property, large surface area to volume ratio and easy functionalization. Homogeneous catalysts immobilized on magnetic nanoparticles (MNPs) surface occupy a unique position due to combining the advantages of both homogeneous and heterogeneous catalysts.17–19 Because of large surface area of nanoparticles, high loadings of catalytically active sites are guaranteed and therefore nanoparticles supported homogeneous catalysts exhibit high catalytic activity and selectivity.16 In addition, the magnetic properties of the Fe3O4 nanoparticles can optimize the operations of separation, recycling, and reuse of the heterogeneous catalyst.20 Hence, these nanoparticles make catalyst recovery more practical and faster than conventional separation methods also reduce solvent consumption. In this way, the important issue of favoring the green chemistry strategy is also covered.21
In the present work, we describe the preparation of an efficient heterogeneous catalyst synthesized by immobilizing manganese porphyrin on functionalized magnetic nanoparticles via the amino propyl linkage. Also, the catalytic activity of the supported catalyst, Fe3O4@SiO2–NH2@MnPor, has been investigated in hydrocarbon oxidation reactions as a recyclable and sustainable nanocatalyst.
2. Experimental
2.1. Materials and methods
All reagents and solvents were purchased from Merck, Fluka or Aldrich chemical companies. Iodosylbenzene22 and tetra-n-butylammonium hydrogen monopersulfate23 were synthesized according to the literature procedure.Scanning electron microscopy (SEM) was carried out on Philips XL30. FT-IR spectra were recorded as KBr pellets using ABB FT-IR spectrophotometer. Measures of pH were carried out by Mettler Toledo S40 SevenMulti™ pH-meter. X-ray diffraction (XRD) pattern was obtained by D4 ENDEAVOR diffractometer (Bruker AXS Inc.) with Cu Kα as a radiation source. Elemental analyses (CHN) were performed using a Heraeus Elemental Analyzer. A Varian (AA220) flame atomic absorption spectrometer (air/acetylene flame) was used for manganese ion determinations. UV-Vis spectra were recorded with a Shimadzu UV-2100 spectrometer. Magnetic measurement of materials was investigated with a vibrating sample magnetometer VSM (Meghnatis Daghigh Kavir Company, Iran) at room temperature. Gas chromatographic (GC) analyses were performed on an Agilent Technologies 6890 N, 19019 J-413 HP-5, capillary 60 m × 250 μm × 1 μm. Brunauer–Emmett–Teller (BET) surface area of the catalyst was measured by Belsorp mini II instrument at −196 °C.
2.2. Porphyrin synthesis and metallation
Porphyrins are a group of heterocyclic macrocycle organic compounds. The porphyrin ring structure is aromatic, with a total of 26 electrons in the conjugated system. Various analyses illustrate that not all atoms of the ring are involved equally in the conjugation.24 One result of this large conjugated system is that porphyrin molecules typically have very intense absorption bands in the visible region. Porphyrins bind metals to form metalloporphyrin complexes. In the present work, the free base, meso-tetrakis(penta-flourophenyl)porphyrin (H2F20TPP or H2TPFPP), was prepared25 and metallated with Mn(OAc)2·4H2O to afford the metalloporphyrin [Mn(TPFPP)OAc].26 The UV-Vis spectrum of homogeneous Mn(TPFPP)OAc was shown in the ESI labeled as Fig. S1.†2.3. Preparation of Fe3O4 magnetic nanoparticles
Fe3O4 nanoparticles were synthesized on the basis of the procedure described previously.19,27,28 In brief, under N2 atmosphere, 5.2 g (19.3 mmol) of FeCl3·6H2O, 2 g (10.0 mmol) of FeCl2·4H2O and 0.85 mL concentrated HCl were dissolved in 25 mL degassed water. This solution was added dropwise at room temperature to 250 mL of NaOH solution (1.5 mol L−1) under N2. The reaction mixture was vigorously stirred for 30 min (1300 rpm). The formed black precipitates were separated using a strong magnetic field (0.5 T magnet) and washed several times with degassed water. Finally, for storage, Fe3O4 nanoparticles were dispersed in 200 mL degassed water under N2. Synthesis of Fe3O4 nanoparticles was approved by XRD.2.4. Synthesis of Fe3O4@SiO2 nanoparticles
In the next step, Fe3O4 MNPs were coated with a thin layer of silica.19,29 One gram of freshly prepared Fe3O4 nanoparticles was added into 30 mL of an aqueous solution of citric acid (0.02 g mL−1), then the pH was adjusted to 5.2 using ammonia, and the mixture was heated to 80–90 °C for 1.5 h. After heating, the pH of the reaction mixture was increased with ammonia to pH = 11 and 1.25 mL of tetraethylorthosilicate (TEOS) dissolved in ethanol (12.5 mL) was added dropwise into the suspension of particles. The mixture was stirred at room temperature for 24 h to allow the base-catalyzed hydrolysis and condensation of TEOS monomers on the nanoparticle surface go to completion. Finally, the dark brown Fe3O4@SiO2 nanoparticles were separated using a 0.5 T magnet and were washed with distilled water and ethanol. Synthesis of Fe3O4@SiO2 nanoparticles was confirmed with X-ray diffraction analysis.2.5. Synthesis of Fe3O4@SiO2–NH2
In a typical reaction, 5 mL of 3-aminopropyltriethoxysilane (APTS) dissolved in 100 mL ethanol were added dropwise to the suspension of one gram of the silica-coated Fe3O4 nanoparticles in 100 mL of distilled water. The pH value of the reaction mixture was increased with KOH to pH = 11 and the reaction mixture was stirred at 70 °C for five hours.15,30 Finally, the brown precipitates were separated using a 0.5 T magnet and were thoroughly washed with distilled water to remove any unbound APTS. Functionalization of Fe3O4@SiO2 nanoparticles was confirmed by FT-IR spectroscopy, and elemental analysis (CHN).2.6. Synthesis of Fe3O4@SiO2–NH2@MnPor nanocatalyst
The process of manganese porphyrin immobilization was conducted by dispersing the synthesized Fe3O4@SiO2–NH2 (0.10 g) in 10 mL of toluene, containing 4.6 × 10−5 mol of metalloporphyrin. The suspension was refluxed and stirred for 24 h under argon atmosphere. The solid was filtered, washed with toluene, CH2Cl2 and ethanol and dried at 60 °C overnight.31,32 The quantity of immobilized manganese porphyrin was determined by measuring the manganese content of the prepared catalyst; Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc], by atomic absorption spectroscopy (AAS).2.7. General oxidation procedure
A typical oxidation reaction using the Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc] nanoparticles as catalyst is described as follows. In a 10 mL round-bottom flask, the prepared catalyst (0.01 g, containing 8 × 10−4 mmol of MnPor), the nitrogenous base of imidazole as the co-catalyst (0.064 mmol), substrate (0.2 mmol) and oxidant (0.4 mmol) were added in order with a molar ratio of {catalyst/imidazole/substrate/oxidant}![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :{1/80/250/500}. The reaction mixture was stirred at room temperature in a tightly closed flask. The progress of the reaction was monitored in different time intervals using gas chromatography (GC). The catalyst nanoparticles were collected at the bottom of the round-bottom flask using a magnet, supernatant carefully decanted and formation of products was examined by GC. The oxidation products were identified by comparison with authentic samples. An internal standard method was used to calculate yields. In the GC experiments, n-decane was used as the internal standard.
:{1/80/250/500}. The reaction mixture was stirred at room temperature in a tightly closed flask. The progress of the reaction was monitored in different time intervals using gas chromatography (GC). The catalyst nanoparticles were collected at the bottom of the round-bottom flask using a magnet, supernatant carefully decanted and formation of products was examined by GC. The oxidation products were identified by comparison with authentic samples. An internal standard method was used to calculate yields. In the GC experiments, n-decane was used as the internal standard.
2.8. Reusability of the catalyst
The reusability and the stability of the catalyst were studied in repeated oxidation reactions. At the end of each reaction, the catalyst was magnetically separated from the reaction mixture, washed with CH2Cl2 and then dried in vacuum at room temperature for 4 h before reusing in the subsequent oxidation reaction. The catalyst was consecutively reused about six times without detectable catalyst leaching or significant loss of activity.3. Results & discussion
As illustrated in Scheme 1, magnetically separable Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc] (1) nanocatalyst was synthesized by multistep procedure. First, superparamagnetic Fe3O4 nanoparticles were prepared using the co-precipitation method. The synthesis of Fe3O4 nanoparticles was followed by coating the surface with a thin silica layer in order to increase the functionality and stability of nanoparticles. For this purpose, the silica-coated MNPs were obtained by basic hydrolysis and condensation of TEOS on the surface of the Fe3O4 nanoparticles. In the next step, Fe3O4@SiO2 nanoparticles have been surface-modified with 3-aminopropyltriethoxysilane (APTS) which introduced –NH2 group on to the surface of support. Eventually, the supported catalyst (1), was prepared by the reaction of Fe3O4@SiO2–NH2 with Mn(TPFPP)OAc. By the procedure of modification of Fe3O4 nanoparticles surface, covalent bonding is a possible approach for this immobilization, because the manganese porphyrin presents the pentafluorophenyl groups at the meso-positioned porphyrin ring, which can be bonded to the functionalized silica by nucleophilic aromatic substitution. This mechanism is described by the reaction of fluorine atoms from the meso porphyrin groups and pendant amino groups from the Fe3O4@SiO2 nanoparticles surface.11,32,33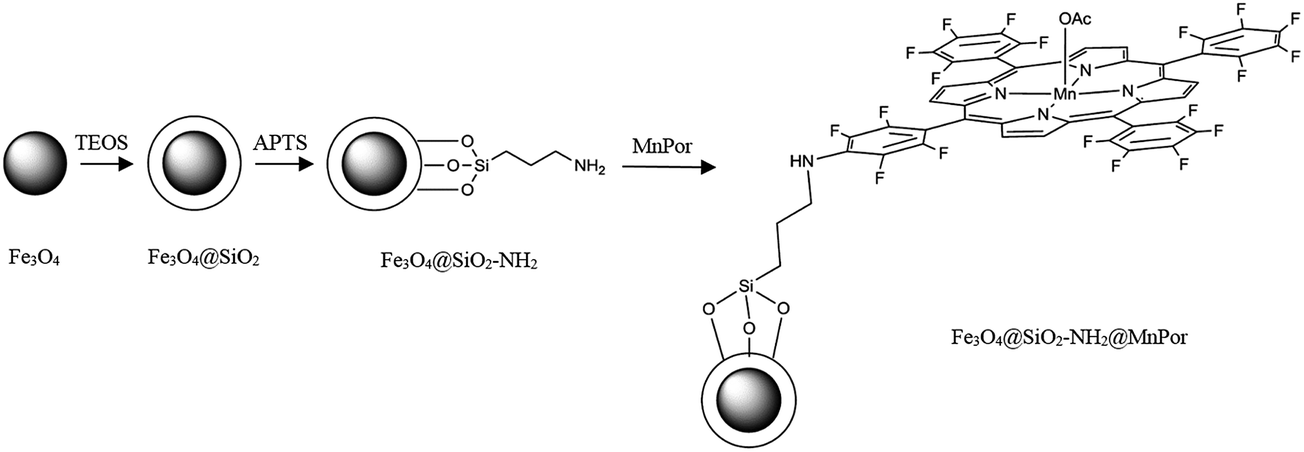 | ||
| Scheme 1 Step-by-step synthesis of the nanocatalyst (1). | ||
3.1. Characterization of the catalyst, Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc]
Fig. 1 shows the XRD patterns of the synthesized compounds as they are shown schematically in Scheme 1. The X-ray diffraction analysis was carried out for the nanoparticles before and after growing a SiO2 shell and after immobilization of the catalyst. In the XRD pattern of the prepared Fe3O4, six characteristic peaks (2θ = 30.6, 35.7, 43.8, 54.2, 57.3, and 63.6), corresponding to (220), (311), (400), (422), (511), and (440) Bragg reflections, respectively, were observed (Fig. 1a). These diffraction peaks are in good agreement with the database in JCPDS file (PCPDFWIN v.2.02, PDF no. 85-1436) and reveal that the resultant nanoparticles were pure Fe3O4 without impurity phases. This XRD pattern is consistent with the pattern previously reported for Fe3O4 samples.19,34 The XRD pattern of Fe3O4@SiO2 core/shell is shown in Fig. 1b. The same characteristic peaks can also be found in this pattern indicating that the crystalline structure of Fe3O4 nanoparticles did not change after the surface modification with silica. The weaker peaks intensity in pattern of Fig. 1b than that of Fig. 1a can be attributed the shielding effect of amorphous silica shell. Also, Fig. 1 shows the XRD patterns of the MNPs–NH2 and MNPs–NH2@MnPor (Fig. 1c and d) with specific peaks and relative intensity, which completely match with the standard Fe3O4 sample.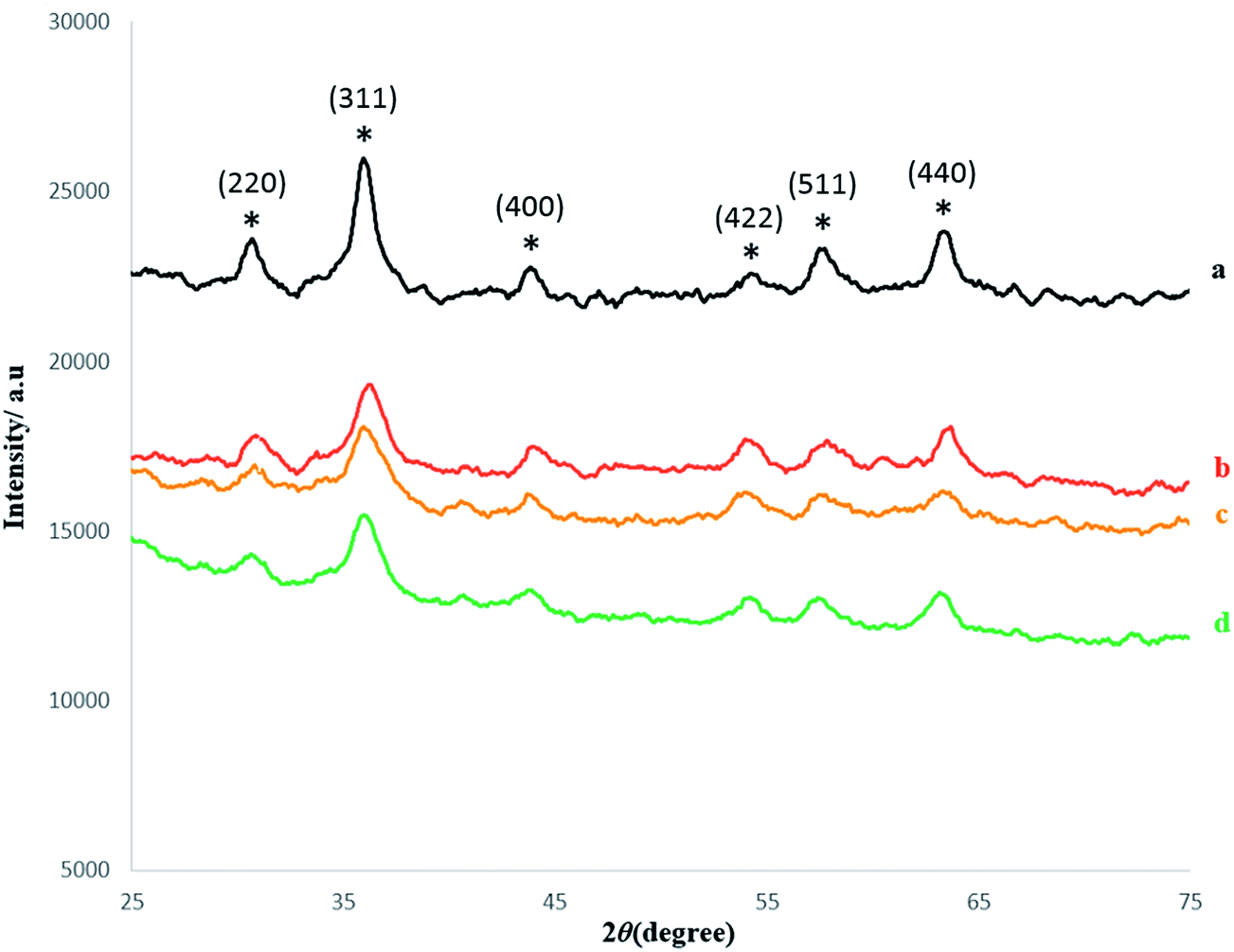 | ||
| Fig. 1 XRD pattern of: (a) Fe3O4, (b) Fe3O4@SiO2 (c) Fe3O4@SiO2–NH2 (d) Fe3O4@SiO2–NH2@MnPor. | ||
Scanning electron microscopy (SEM) image of Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc] catalyst clearly depicts a smooth morphology of the nanocatalyst; particles were well distributed with diameters around 27 nm and rather high surface area (Fig. 2). Brunauer–Emmett–Teller (BET) analysis of the nanocatalyst was performed and the obtained N2 adsorption–desorption isotherm is depicted in Fig. S2† which is located in ESI. The BET analysis is the most common method for determining surface areas from nitrogen adsorption isotherms.35 The measured BET surface area is 81.2 m2 g−1 for the nanocatalyst (1). Therefore, the relatively high surface area of nanoparticles is beneficial for high loading of catalyst during the complex anchoring step.
 | ||
| Fig. 2 The scanning electron microscopy image of Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc]. | ||
Because of the presence of the magnetic core inside, MNPs cannot be analyzed by means of solid state NMR spectroscopy. Therefore amino functionalized nanoparticles (Fe3O4@SiO2–NH2) were systematically characterized by FT-IR and elemental analysis. Fig. 3 shows the FT-IR spectra of the core–shell magnetic nanoparticles (Fe3O4@SiO2) after being functionalized by APTS. The FT-IR spectrum (Fig. 3a) represents significant absorption bands at about 586 and 632 cm−1 which corresponds to Fe–O vibration modes of Fe3O4. Furthermore, a strong absorption band at around 1068 cm−1 (between 1000 and 1200 cm−1) and a band at 794 cm−1, which can be assigned as vibration modes of Si–O–Si,36,37 indicate that the Fe3O4 core is successfully coated by silica shell. Another bands at around 1622 and 3427 cm−1 are associated with the vibrations of absorbed water molecules.38
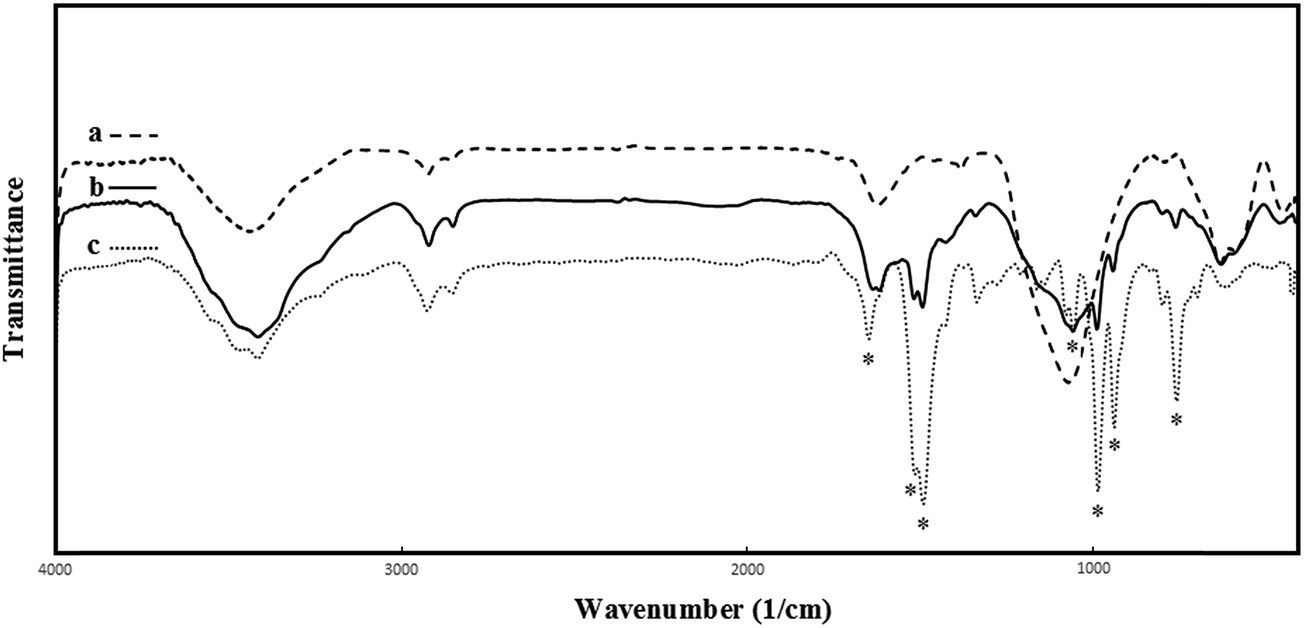 | ||
| Fig. 3 FT-IR spectra of: (a) Fe3O4@SiO2–NH2 (b) Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc], (c) Mn(TPFPP)OAc. | ||
Additionally, the characteristic peaks at 2854 and 2923 cm−1 ascribed to the C–H stretching vibration of the propyl group in the pendant APTS can be clearly observed in the FT-IR spectrum of Fe3O4@SiO2–NH2, which confirms that APTS molecules have been bonded successfully to the surface of the silica-coated MNPs.39 This conclusion is further supported by the elemental analysis which gave the percentages of C, H, and N to be 5.10%, 1.54%, and 1.53%, respectively. The frequency of N–H asymmetric and symmetric stretching vibrations of the amine group fall in the 3300–3400 cm−1 range and are obscured by the water band.
Furthermore, immobilization of metalloporphyrin on APTS-coated magnetic nanoparticles was exhibited by FT-IR spectroscopy. Comparing FT-IR spectra of Fe3O4@SiO2–NH2@MnPor (Fig. 3b) and Mn-porphyrin (Fig. 3c) revealed that signals at about 759, 941, 987, 1492, 1517 and 1649 cm−1 corresponding to the vibration modes of pure metalloporphyrin are present in the FT-IR spectra of Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc] nanoparticles. The loading of manganese porphyrin complex was 0.08 mmol g−1 of the supported nanocatalyst, determined by AAS analysis.
The presence of MnPor on functionalized magnetic nanoparticles was analyzed by means of UV-Vis spectroscopy (Fig. 4). No absorption band was observed in the UV-Vis spectra of Fe3O4@SiO2–NH2 (Fig. 4b, dash line). However, the Soret and Q bands at 475 and 576 nm respectively were appeared after immobilization process of Mn(TPFPP)OAc on the surface (Fig. 4a). In this way, both of the FT-IR and UV-Vis spectroscopy provide evidences for anchoring of the manganese porphyrin on APTS-coated Fe3O4 NPs.
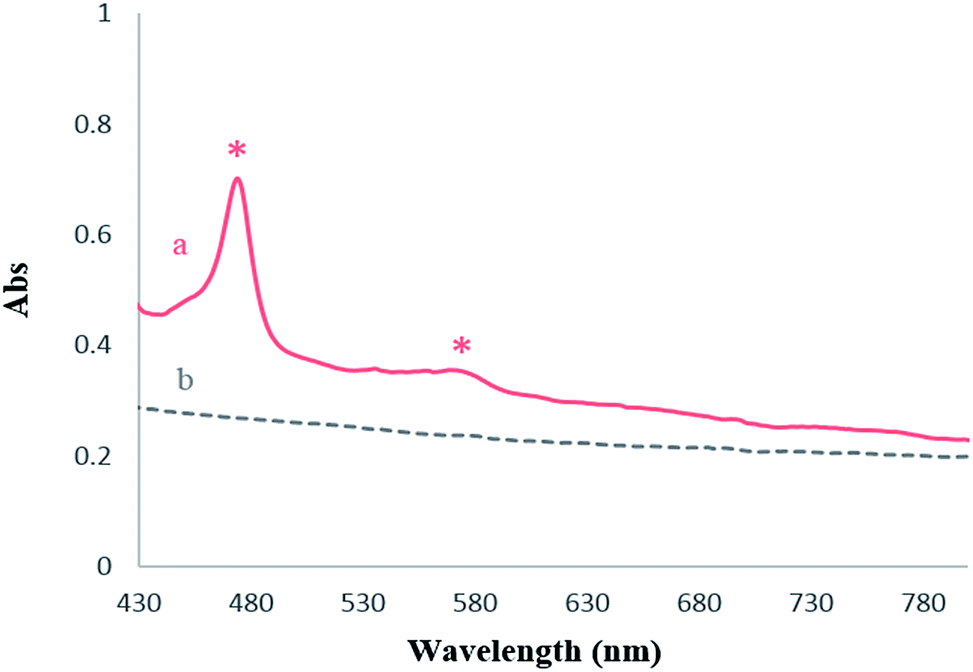 | ||
| Fig. 4 UV-Vis spectra of: (a) Fe3O4@SiO2–NH2, (b) Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc]. | ||
The magnetic properties of uncoated Fe3O4 nanoparticles, Fe3O4@SiO2, and the Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc] catalyst were characterized by a vibrating sample magnetometer (VSM) under an applied field of −10000 to 10000 Oe at room temperature (Fig. 5). According to the magnetization curves, the reduced saturation magnetization values observed for the functionalized nanoparticles (Fig. 5b and c) as compared with pure Fe3O4 nanoparticles (Fig. 5a) were due to the diamagnetic surface coating and surface functionalization of Fe3O4 nanoparticles.40,41 However, both in the case of the core–shell MNPs and of the final nanocatalyst, such values are still large enough for nanoparticles to be easily separated by an external magnet.
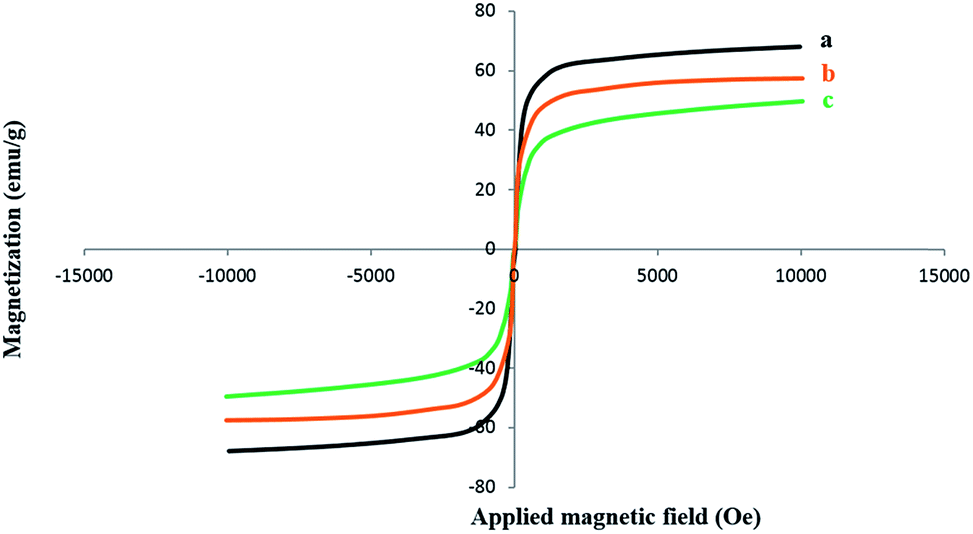 | ||
| Fig. 5 Magnetization curves of: (a) Fe3O4 (b)Fe3O4@SiO2 (c) Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc]. | ||
3.2. Catalytic experiments
The present work reports the catalytic activity of Mn(TPFPP)OAc supported on APTS-coated Fe3O4@SiO2 nanoparticles; Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc] (1), in alkane oxidation in the presence of two different oxidants; tetra-n-butylammonium hydrogen monopersulfate (n-Bu4NHSO5) and iodosylbenzene (PhIO). Catalytic performance of heterogeneous catalyst (1) in alkene epoxidation with n-Bu4NHSO5 was also investigated as shown in Scheme 2.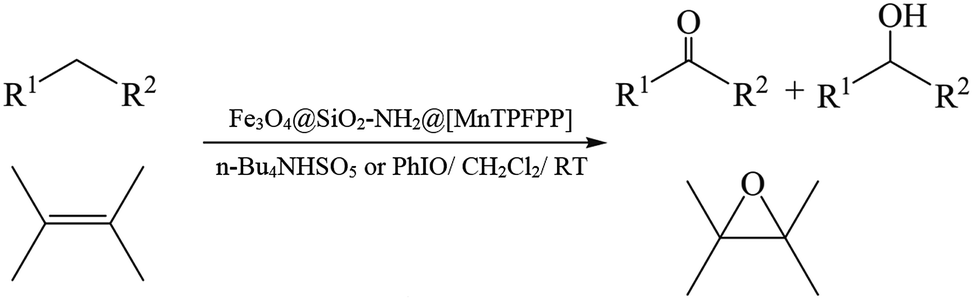 | ||
| Scheme 2 Hydroxylation of alkanes and epoxidation of alkenes catalyzed by Fe3O4@SiO2–NH2@[MnTPFPP] (1). | ||
It is believed that the employment of imidazoles in metalloporphyrin systems for mimicking the axial coordination function of cytochrome-P450 has led to remarkable improvement in oxidation reactions.42,43 According to the co-catalytic role of axial base in biomimetic oxidations catalyzed with manganese porphyrins,44,45 imidazole nitrogenous base was employed as axial ligand in the mentioned reactions. Imidazole with the ability of σ-donating and π-donating, mainly exhibited higher activity as a co-catalyst, compared to other nitrogenous bases.
The oxidation reactions were carried out with molar ratio of {catalyst/imidazole/substrate/oxidant}:{1/80/250/500} at room temperature in CH2Cl2 as the reaction medium.
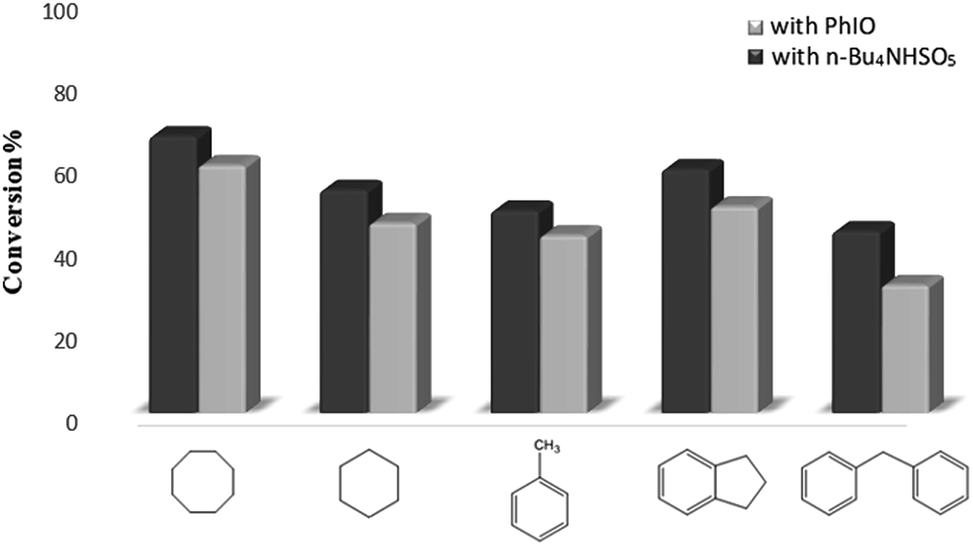 | ||
| Fig. 6 Oxidation of alkanes catalyzed by Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc] in the presence of two different oxidants: n-Bu4NHSO5 and PhIO. | ||
This heterogenized catalytic system converts different alkanes to their corresponding alcohols and aldehyde/ketones (Table 1). The oxidation of cyclooctane, cyclohexane, toluene, indane and diphenylmethane in dichloromethane solution at room temperature catalyzed by immobilized Mn-porphyrin gives respectively cyclooctanone, cyclohexanone, benzaldehyde, 1-indanone and benzophenone as the main product, as well as smaller amounts of cyclooctanol, cyclohexanol, benzyl alcohol and 1-indanol. In addition; to study the influence of time in the oxidation process, the reactions were carried out in two different time intervals. It can be observed that Fe3O4@SiO2–NH2@[Mn(TPFPP)OAc] is an efficient catalyst for oxidation of alkanes in the presence of n-Bu4NHSO5. Therefore, the results of this study show better catalytic performance of the heterogonous catalyst using n-Bu4NHSO5 as oxidant in comparison with PhIO in oxidation reactions both in terms of selectivity and conversion levels.
| Entry | Alkane | Conversiona,b% | Selectivity to aldehyde/ketonee% | TONf |
|---|---|---|---|---|
| a Reaction conditions: alkane (0.2 mmol), n-Bu4NHSO5 (0.4 mmol), catalyst (8 × 10−4 mmol), CH2Cl2 (1 mL), reaction time: 20 h.b Determined by GC.c Values in parentheses were obtained after 4 h.d Values in brackets were obtained in the presence of PhIO.e Selectivity to (aldehyde/ketone) = ((aldehyde/ketone)/((aldehyde/ketone) + alcohol)) × 100.f TON = (mmol of product)/mmol of catalyst. | ||||
| 1 |  |
71 (67)c [64 (60)c]d | 62 [60]d | 177.5 |
| 2 | 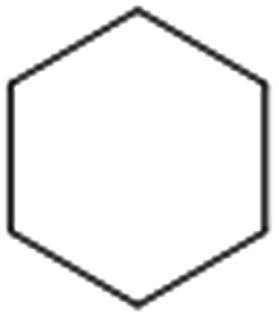 |
55 (54)c [48 (46)c]d | 80 [76]d | 137.5 |
| 3 | 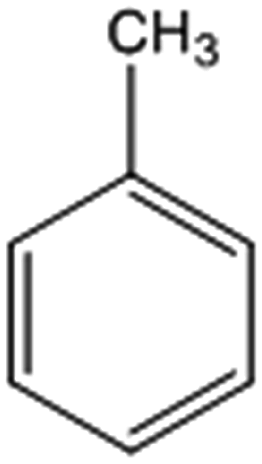 |
52 (49)c [44 (43)c]d | 95 [89]d | 130 |
| 4 | 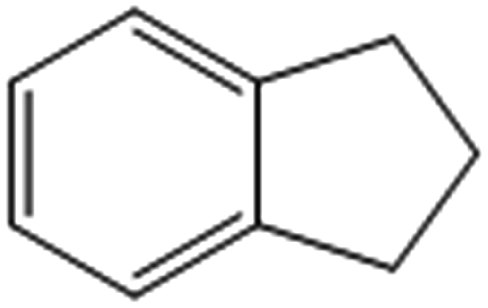 |
65 (59)c [57 (50)c]d | 73 [70]d | 162.5 |
| 5 | 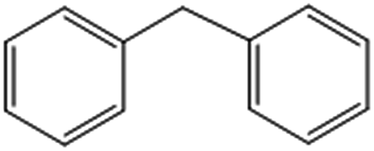 |
47 (44)c [36 (31)c]d | 100 [100]d | 117.5 |
A cage-controlled radical mechanism, the “oxygen rebound” mechanism, has been proposed and discussed for the manganese(III) porphyrin-catalyzed hydroxylation of alkanes.48–50 This process involves two steps: firstly, hydrogen atom abstraction by the active specie MnV(O)P generates a solvent-caged alkyl radical and a hydroxymanganese(IV) species (Scheme 3, pathway A); in the second step, a subsequent ‘in cage’ reaction leads to the formation of the alcohol and the MnPor is recovered. However, in competition with the second step, the carbon radicals can escape from the cage (Scheme 3, pathway B), thus triggering radical processes, with the production of ketone products.51,52
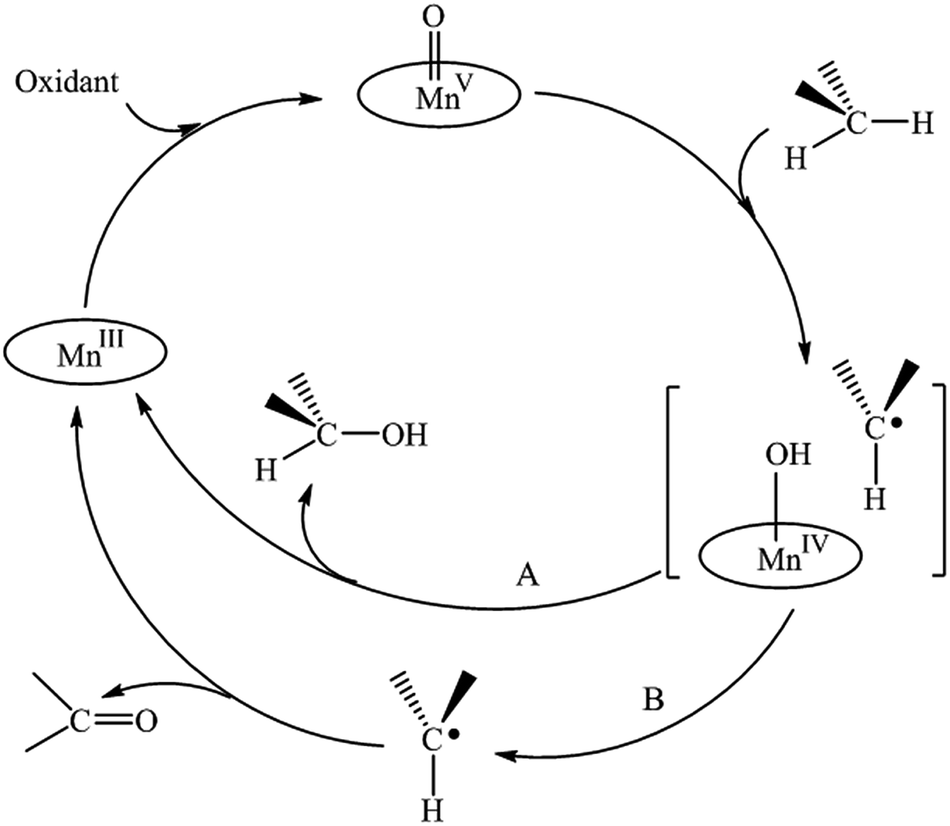 | ||
| Scheme 3 Mechanism of the MnPor-catalyzed oxidation of alkanes. | ||
| Entry | Alkene | Conversiona,b% | Selectivity to epoxide% | TONf |
|---|---|---|---|---|
| a Reaction conditions: alkene (0.2 mmol), n-Bu4NHSO5 (0.4 mmol), catalyst (8 × 10−4 mmol), CH2Cl2 (1 mL), reaction time: 20 h.b Determined by GC.c Values in parentheses were obtained after 3 h.d The by-product is allylic ketone.e The by-product is benzaldehyde.f TON = (mmol of product)/mmol of catalyst. | ||||
| 1 | 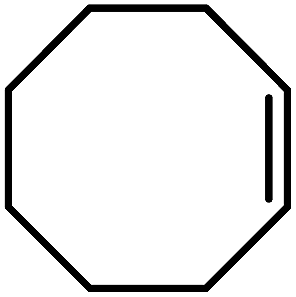 |
97 (85)c | 100 | 242.5 |
| 2 | 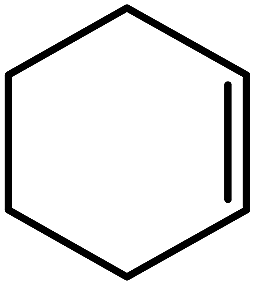 |
93d (83)c,d | 94 | 232.5 |
| 3 | 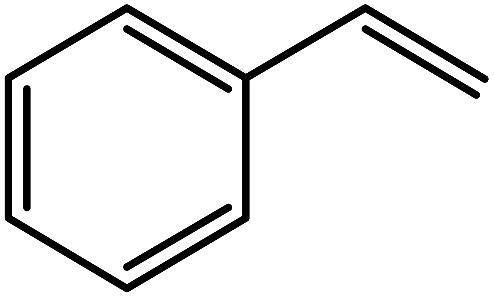 |
94e (81)c,e | 92 | 235 |
| 4 |  |
48 (37)c | 100 | 120 |
| 5 |  |
56 (43)c | 100 | 140 |
For alkene epoxidation, there are some mechanism reports of parallel reactions occurring besides the concerted epoxidation mechanism (Scheme 4, pathway A). In these mechanisms, the active specie MnV(O)P forms a charge transfer complex with the alkene.1,57–59 As shown in Scheme 4, two processes can occur: (B) the charge transfer complex generates a radical intermediate, that will attack the alkene generating the epoxidized product, or (C) the charge transfer complex generates a solvent-caged alkyl radical, which favors the electron transfer from the alkene to the MnV(O)P, followed by the formation of a carbocation and subsequent oxygen transfer, thus generating the epoxidized product.48,60 It should be mentioned whenever the reaction goes through each of the radical pathways (B and/or C), a variety of products may be readily yielded. According to the negligible difference of conversion and epoxidation yield as shown in Table 2, epoxide seems to be the sole main product. Therefore a concerted mechanism (pathway A) may play a role here for the reaction progress.
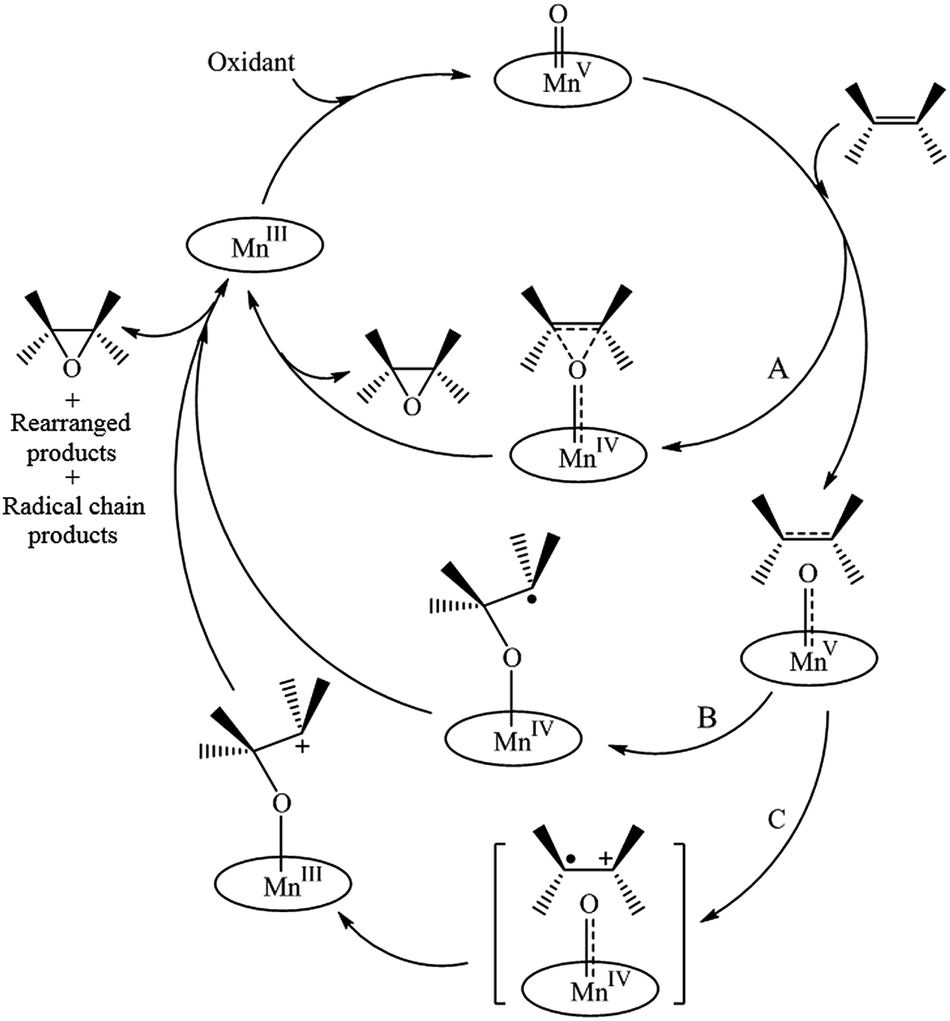 | ||
| Scheme 4 Mechanism of the MnPor-catalyzed oxidation of alkenes. | ||
Comparison of the catalytic performances presented in this work with those of other similar studies61–67 reveals that our catalytic system is superior to some of the previously reported catalysts in terms of catalytic activity (TON) and reaction conditions. The high turnover number of our catalyst compared to recently reported protocols61,62 makes the Fe3O4@SiO2–NH2@[MnTPFPP] more attractive for oxidation of hydrocarbons. In other words, compared with these nonporphyrin catalysts, our catalytic system exhibits high catalytic activity in term of high TON. According to the molar ratio of 1/250/500 for catalyst/substrate/oxidant which is carried out in this work, the amount of catalyst which is used in each run (0.01 g, containing 8 × 10−4 mmol of MnPor) is lower than that of previous works.61–66
It is observed that in several articles,62,65 performance of the catalyst in oxidation of various substrates was inspected under reflux, while the oxidation reactions were carried out at room temperature in this work. It should be mentioned that the insertion of an oxygen atom within the C–H of a saturated carbon atom is one of the most difficult reactions to achieve with a catalyst at room temperature.
Our heterogenized catalytic system, can be recovered and reused in comparison with the homogeneous complexes.61–64 Metal complexes are stabilized upon immobilization on solid supports because of the support environment. Also, the formation of inactive dimer, the major drawback of homogeneous catalyst, is prevented by site isolation on the surface. Moreover, non-magnetic heterogeneous systems66,67 require filtration and centrifugation steps to recover the catalysts leading to loss of catalytic activity during consecutive cycles. Therefore, the magnetically supported nanocatalyst, the present work, can be conveniently separated from reaction solution with a magnet and reused several times without significant loss of activity.
4. Conclusions
This work describes the covalent immobilization of Mn-porphyrin, 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin manganese(III) acetate Mn(TPFPP)OAc, onto magnetic nanoparticles covered with amino functionalized silica. This magnetically separable metalloporphyrin was applied as an efficient retrievable heterogeneous nanocatalyst for the oxidation of a variety of alkanes and alkenes. In other words, this heterogenized catalytic system could join the catalytic properties of the manganese porphyrin with the magnetic properties of Fe3O4 nanoparticles. The results show that the dispersion of these superparamagnetic nanoparticles, which can be reversibly controlled by applying an external magnetic field, introducing them as the recyclable support matrix in the catalytic oxidations. Because of the relatively high surface area of these nanoparticles, high loadings of catalytically active sites are guaranteed and therefore high catalytic activity is provided for the supported metalloporphyrin catalyst.Furthermore, the separation and reuse of the magnetic Fe3O4 nanoparticles were very effective and economical. Leaching and recycling experiments revealed that nanocatalyst is already recyclable several times without loss of activity and magnetic properties. The attractive features of this catalytic system such as easy work up, noticeable reusability and mild reaction conditions make it particularly suitable for hydrocarbon oxidation reactions.
Acknowledgements
We would like to appreciate the Sharif University of Technology Research Council for research funding of this project.References
- P. R. Ortiz de Montellano, Cytochrome P450: Structure, Mechanism, and Biochemistry, Plenum Press, New York, 2004 Search PubMed.
- D. Dolphin, T. G. Traylor and L. Y. Xie, Acc. Chem. Res., 1997, 30, 251–259 CrossRef CAS.
- J. P. Collman, X. Zhang, V. J. Lee, E. S. Uffelman and J. I. Brauman, Science, 1993, 261, 1404–1411 CAS.
- R. A. Sheldon and B. Meunier, Biomimetic Oxidations Catalysed by Transition Metal Complexes, Imperial College Press, London, 2000 Search PubMed.
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed.
- E. Brule and Y. R. De Miguel, Org. Biomol. Chem., 2006, 4, 599–609 CAS.
- M. A. Schiavon, Y. Iamamoto, O. R. Nascimento and M. D. D. Assis, J. Mol. Catal. A: Chem., 2001, 174, 213–222 CrossRef CAS.
- N. Bizaia, E. H. de Faria, G. P. Ricci, P. S. Calefi, E. J. Nassar, K. A. D. F. Castro, S. Nakagaki, K. J. Ciuffi, R. Trujillano, M. A. Vicente, A. Gil and S. A. Korili, ACS Appl. Mater. Interfaces, 2009, 1, 2667–2678 CAS.
- A. K. Rahiman, S. Sreedaran, K. S. Bharathi and V. Narayanan, J. Porous Mater., 2010, 17, 711–718 CrossRef.
- G. S. Machado, K. A. D. de Freitas Castro, F. Wypych and S. Nakagaki, J. Mol. Catal. A: Chem., 2008, 283, 99–107 CrossRef CAS.
- M. Halma, K. A. D. de Freitas Castro, V. Prévot, C. Forano, F. Wypych and S. Nakagaki, J. Mol. Catal. A: Chem., 2009, 310, 42–50 CrossRef CAS.
- S. Evans and J. R. Lindsay Smith, J. Chem. Soc., Perkin Trans. 2, 2001, 174–180 RSC.
- J. Safari and L. Javadian, RSC Adv., 2014, 4, 48973–48979 RSC.
- N. Zohreh, S. H. Hosseini, A. Pourjavadi and C. Bennett, RSC Adv., 2014, 4, 50047–50055 RSC.
- H. Keypour, M. Balali, M. M. Haghdoost and M. Bagherzadeh, RSC Adv., 2015, 5, 53349–53356 RSC.
- S. Shylesh, W. R. Thiel and V. Schunemann, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- K. V. S. Ranganath, J. Kloesges, A. H. Schäfer and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7786–7789 CrossRef CAS PubMed.
- K. V. S. Ranganath and F. Glorius, Catal. Sci. Technol., 2011, 1, 13–22 CAS.
- M. Bagherzadeh and A. Mortazavi-Manesh, J. Coord. Chem., 2015, 68, 2347–2360 CrossRef CAS.
- A. Schätz, O. Reiser and W. J. Stark, Chem.–Eur. J., 2010, 16, 8950–8967 CrossRef PubMed.
- N. Saadatjoo, M. Golshekan, S. Shariati, H. Kefayati and P. Azizi, J. Mol. Catal. A: Chem., 2013, 377, 173–179 CrossRef CAS.
- H. Saltzman and J. G. Sharefkin, Org. Synth., 1973, 5, 658–663 Search PubMed.
- S. Compestrini and B. Meunier, Inorg. Chem., 1992, 31, 1999–2006 CrossRef.
- A. S. Ivanova and A. I. Boldyrev, Org. Biomol. Chem., 2014, 12, 6145–6150 Search PubMed.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827–836 CrossRef CAS.
- K. M. Kadish, C. Araullo-McAdams, B. C. Han and M. M. Franzen, J. Am. Chem. Soc., 1990, 112, 8364–8368 CrossRef CAS.
- X. Q. Liu, Z. Y. Ma, J. M. Xing and H. Z. Liu, J. Magn. Magn. Mater., 2004, 270, 1–6 CrossRef CAS.
- M. Bagherzadeh, M. M. Haghdoost and A. Shahbazirad, J. Coord. Chem., 2012, 65, 591–601 CrossRef CAS.
- S. Campelj, D. Makovec and M. Drofenik, J. Magn. Magn. Mater., 2009, 321, 1346–1350 CrossRef CAS.
- M. Bagherzadeh, M. M. Haghdoost, F. Matlobi-Moghaddam, B. Koushki-Foroushani, S. Saryazdi and E. Payab, J. Coord. Chem., 2013, 66, 3025–3036 CrossRef CAS.
- F. Wypych, A. Bail, M. Halma and S. Nakagaki, J. Catal., 2005, 234, 431–437 CrossRef CAS.
- P. Battioni, J. F. Bartoli, D. Mansuy, Y. S. Byun and T. G. Traylor, J. Chem. Soc., Chem. Commun., 1992, 15, 1051–1053 RSC.
- M. D. Assis and J. R. Lindsay Smith, J. Chem. Soc., Perkin Trans. 2, 1998, 2221–2226 RSC.
- J. Wang, Q. W. Chen, C. Zeng and B. Y. Hou, Adv. Mater., 2004, 16, 137–140 CrossRef CAS.
- S. K. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552–8556 CrossRef PubMed.
- K. D. Kim, S. S. Kim, Y. H. Choa and H. T. Kim, J. Ind. Eng. Chem., 2007, 13, 1137–1141 CAS.
- J. H. Cai, J. W. Huang, P. Zhao, Y. J. Ye, H. C. Yu and L. N. Ji, J. Sol-Gel Sci. Technol., 2009, 50, 430–436 CrossRef CAS.
- C. Yuan, Z. Huang and J. Chen, Catal. Lett., 2011, 141, 1484–1490 CrossRef CAS.
- P. Das, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, J. Mater. Sci., 2009, 44, 2865–2875 CrossRef CAS.
- Z. U. Rahman, Y. Dong, L. Su, Y. Ma, H. Zhang and X. Chen, Chem. Eng. J., 2013, 222, 382–390 CrossRef.
- M. Masteri-Farahani and N. Tayyebi, J. Mol. Catal. A: Chem., 2011, 348, 83–87 CrossRef CAS.
- Z. Gross and S. Ini, J. Org. Chem., 1997, 62, 5514–5521 CrossRef CAS.
- T. Matsui, S. Oyaki and Y. Watanabe, J. Am. Chem. Soc., 1999, 121, 9952–9957 CrossRef CAS.
- D. Mohajer, G. Karimipour and M. Bagherzadeh, New J. Chem., 2004, 28, 740–747 RSC.
- A. Aghabali and N. Safari, J. Porphyrins Phthalocyanines, 2010, 14, 335–342 CrossRef CAS.
- J. L. McLain, J. Lee, J. T. Groves and B. Meunier, Biomimetic Oxidations Catalyzed by Transition Metal Complexes, Imperial College Press, London, 2000, pp. 91–170 Search PubMed.
- C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC.
- S. P. de Visser and W. Nam, in Handbook of Porphyrin Science: Catalysis and Bio-Inspired Systems, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing, Singapore, 2010, pp. 85–140 Search PubMed.
- J. T. Groves, W. J. Kruper and R. C. Haushalter, J. Am. Chem. Soc., 1980, 102, 6375–6377 CrossRef CAS.
- R. J. Balahura, A. Sorokin, J. Bernadou and B. Meunier, Inorg. Chem., 1997, 36, 3488–3492 CrossRef CAS PubMed.
- J. Bernadou and B. Meunier, Chem. Commun., 1998, 20, 2167–2173 RSC.
- J. A. Smegal and C. L. Hill, J. Am. Chem. Soc., 1983, 105, 3515–3521 CrossRef CAS.
- M. R. dos Santos, J. R. Diniz, A. M. Arouca, A. F. Gomes, F. C. Gozzo, S. M. Tamborim, A. L. Parize, P. A. Z. Suarez and B. A. D. Neto, ChemSusChem, 2012, 5, 716–726 CrossRef CAS PubMed.
- W. S. D. Silva, A. A. M. Lapis, P. A. Z. Suarez and B. A. D. Neto, J. Mol. Catal. B: Enzym., 2011, 68, 98–103 CrossRef CAS.
- P. A. Z. Suarez, M. S. C. Pereira, K. M. Doll, B. K. Sharma and S. Z. Erhan, Ind. Eng. Chem. Res., 2009, 48, 3268–3270 CrossRef CAS.
- P. Adao, J. C. Pessoa, R. T. Henriques, M. L. Kuznetsov, F. Avecilla, M. R. Maurya, U. Kumar and I. Correia, Inorg. Chem., 2009, 48, 3542 CrossRef CAS PubMed.
- R. D. Arasasingham, G. X. He and T. C. Bruice, J. Am. Chem. Soc., 1993, 115, 7985–7991 CrossRef CAS.
- J. T. Groves and M. K. Stern, J. Am. Chem. Soc., 1987, 109, 3812–3814 CrossRef CAS.
- W. Nam, I. Kim, M. H. Lim, H. J. Choi, J. S. Lee and H. G. Jang, Chem.–Eur. J., 2002, 8, 2067–2071 CrossRef CAS.
- T. G. Traylor, Y. Iamamoto and T. Nakano, J. Am. Chem. Soc., 1986, 108, 3529–3531 CrossRef CAS.
- M. Amini, M. Bagherzadeh, Z. Moradi-Shoeili, D. M. Boghaei, A. Ellern and L. K. Woo, J. Coord. Chem., 2013, 66, 464–472 CrossRef CAS.
- T. Heikkilä, R. Sillanpää and A. Lehtonen, J. Coord. Chem., 2014, 67, 1863–1872 CrossRef.
- M. Amini, M. Bagherzadeh, B. Eftekhari-Sis, A. Ellern and L. K. Woo, J. Coord. Chem., 2013, 66, 1897–1905 CrossRef CAS.
- M. Bagherzadeh, M. Amini, A. Ellern and L. K. Woo, Inorg. Chem. Commun., 2012, 15, 52–55 CrossRef CAS.
- M. Zare, Z. Moradi–Shoeili, M. Bagherzadeh, S. Akbayrak and S. Özkar, New J. Chem., 2015, 1–8 Search PubMed.
- S. Rayati, S. Zakavi, P. Jafarzadeh, O. Sadeghi and M. M. Amini, J. Porphyrins Phthalocyanines, 2012, 16, 260–266 CrossRef CAS.
- S. Rayati, P. Jafarzadeh and S. Zakavi, Inorg. Chem. Commun., 2013, 29, 40–44 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02123a |
| This journal is © The Royal Society of Chemistry 2016 |