
PdCl2 immobilized on metal–organic framework CuBTC with the aid of ionic liquids: enhanced catalytic performance in selective oxidation of cyclohexene†
Abstract
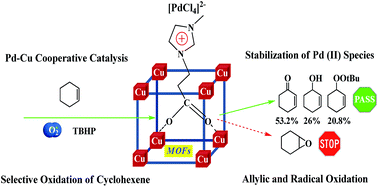
Metal–organic framework CuBTC was employed as a support to immobilize PdCl2 with the aid of ionic liquids (ILs). The as-synthesized PdCl2-ILs/CuBTC catalyst was studied in selective oxidation of cyclohexene with molecular oxygen as an oxidant and TBHP as an initiator. It is found that the allylic oxidation and radical oxidation processes are main reaction pathways. More importantly, the enhancement of catalytic activity in the oxidation of cyclohexene is observed over the PdCl2-IL/CuBTC catalyst due to Pd–Cu cooperative catalysis. Furthermore, ionic liquids could have a very favourable role in the stabilization of Pd(II) species and improvement of catalyst reusability.
 Please wait while we load your content...
Please wait while we load your content...

