
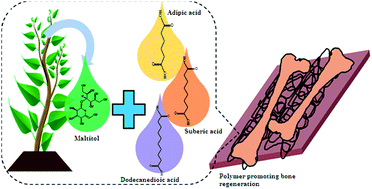
Maltitol-based biodegradable polyesters with tailored degradation and controlled release for bone regeneration†
 b
and
b
and
Abstract
Despite extensive research performed in the area of drug delivery and tissue engineering, the search for a perfect biomaterial remains an ongoing process. In an effort to find this material, novel maltitol-based polyesters using three different dicarboxylic acids (DCAs; adipic acid, dodecanedioic acid and suberic acid) were synthesized and their properties were investigated. The chemical structure of the polymers was confirmed using Fourier transform infrared and proton nuclear magnetic resonance spectroscopies. Thermal characterization revealed that the polymers were amorphous and that the glass transition temperature decreased with an increase in the chain length and molar ratio of maltitol : DCAs. Mechanical studies showed that the moduli of these polymers were comparable to those of the components of the skeletal system. Contact angle goniometry confirmed that the hydrophobicity of the polymers increased with increase in chain length and the molar ratio of maltitol : DCAs. The polymer degradation followed first order kinetics whereas dye release from these polymers followed zero order kinetics. Both the degradation and dye release studies demonstrated that the degradation and release decreased with increase in chain length and molar ratio of maltitol : DCA. The degradation and dye release can be modulated based on the chain length and the molar ratio of acids. Preliminary cytocompatibility studies showed that these polymers were cytocompatible. Mineralization studies revealed that these polymers showed increased mineralization when compared to results obtained with controls. Thus, this family of polyesters can serve as effective biomaterials for bone tissue engineering with tunable degradation and controlled release properties.
 Please wait while we load your content...
Please wait while we load your content...

