
Highly enhanced leukemia therapy and oral bioavailability from a novel amphiphilic prodrug of cytarabine†
Jing Liu‡
a,
Jing Liu‡ab,
Dujuan Zhaoa,
Naxin Maa and
Yuxia Luan*a
aSchool of Pharmaceutical Science, Shandong University, 44 West Wenhua Road, Jinan, Shandong Province 250012, P. R. China. E-mail: yuxialuan@sdu.edu.cn; Fax: +86 531 88382548; Tel: +86 531 88382007
bChia Tai Tian Qing Pharmaceutical Group Co., Ltd., Lianyungang, Jiangsu Province 222000, P. R. China
First published on 24th March 2016
Abstract
Cytarabine (1-β-D-arabinofuranosylcytosine, Ara-C), an attractive medicinal treatment for myeloblastic leukemia, is subject to low oral bioavailability due to its weak membrane permeability and lipophilicity as well as its poor metabolic stability. Based on these considerations, we proposed a chemical linkage of cytarabine with lauric acid (LA), a long-chain fatty acid with 12 carbons, leading to a new prodrug, LA–Ara. The NH2 group of Ara-C is protected by conjugation and thus cannot not be deactivated by deamination; moreover, notably high liposolubility and penetrability were obtained. When dispersed in water, this new amphiphilic molecule can adopt a nanofiber configuration. It was found that LA–Ara molecules are stable in artificial biological media, indicating that the prodrug could be administrated orally. MTT assays on HL60 and K562 cells were performed, and the significantly higher cytotoxicity compared to the pure drug suggested that the prodrug has conspicuously superior antiproliferative activity. Following oral administration, the elimination half-life and bioavailability of the LA–Ara group dramatically increased to 6.6- and 32.8-fold of those of free Ara-C, respectively. It appears that this new prodrug could be an effective oral alternative treatment to Ara-C and supply a promising therapy index for leucocythemia.
1. Introduction
1-(β-D-Arabinofuranosyl) cytosine (cytarabine, Ara-C) is a potent therapeutic agent for the treatment of both acute and chronic myeloid leukemias.1,2 It also plays a vital role in the therapy of solid tumors in combination with other anticancer drugs.3 Via intracellular phosphorylation, Ara-C can be continuously converted to its major active metabolite, 1-β-D-arabinofuranoside 5′-triphosphate (Ara-CTP), whose effects include both inhibition of DNA polymerase and incorporation into DNA.4,5 Subsequently, the interruption of DNA replication triggers apoptosis in cancer cells and inhibits tumor growth. Although it is the cornerstone for myeloid leukemia therapy, Ara-C suffers from several limitations, which limit its clinical utility. The absolute water-solubility of Ara-C prevents the molecules from penetrating cells and gastrointestinal membranes. Moreover, rapid deamination to the biologically inactive 1-D-arabinofuranosyluracil (Ara-U) by cytidine deaminase in intestinal and hepatic cells reduces the circulation time, resulting in an extremely short plasma half-life and low oral bioavailability (about 20%).6 Due to these initial drawbacks, intravenous infusion with a complex and precise dosing schedule is usually adopted as the clinical standard administration to maintain a constant plasma level in humans.7,8 Therefore, clinical applications and patient tolerability of Ara-C are severely hindered, and it is highly imperative to develop an alternative oral administration for this drug.Prodrugs are a series of conjugates which are therapeutically effective after being metabolized to the bioactive parent component in vivo. As an important strategy to address the shortcomings of promising candidates, prodrugs have been widely utilized to improve the efficacy of existing anticancer agents.9 For example, gemcitabine, a nucleoside analogue which has the same restrictions as Ara-C, was conjugated to squalene, generating a lipophilic prodrug with significantly enhanced pharmacokinetic behavior and antitumor activity compared with the unmodified drug.10 Somewhat analogous findings have also been reported for N-acyl derivatives of gemcitabine.11,12 The first prodrug which had clearly superior activities to pure Ara-C was adamantoyl cytarabine;13 since that time, a host of derivatives of Ara-C have been synthesized. Generally, alteration occurs either by substitution at the amine group of the cytosine base or at the 5′-hydroxyl group of the arabinose sugar, producing products with compromised activity.14–16 Considering the obstacles to the clinical utility of Ara-C, the major impetus to the present work was the development of orally available Ara-C derivatives; few such derivatives have led to approved prodrugs to date. N4-L-valyl-Ara-C was synthesized by introducing an amino acid to the 4′-NH2 group, which was expected to increase the oral absorption via the carrier-mediated transport pathway; however, the bioavailability was only 4% after oral administration in rats.17,18 A series of 5′-amino acid ester derivatives targeting intestinal PepT1 have been considered as hopeful prospects to address the disadvantages of Ara-C. Although the absorption of the drug was passable, there was still a reasonable quantity of inactive Ara-U due to the exposed NH2 group.19 In our previous study, we synthesized a new di-cytarabine prodrug, Ara-R-Ara, and a mono-amide prodrug, OA-Ara. Both these compounds demonstrated excellent anticancer efficiency in in vitro cytotoxicity tests. Unfortunately, the pharmaceutic behavior of this type of aliphatic ramification is yet unclear.20,21
Thus, in this work, lauric acid (LA) was covalently attached to the 4′-NH2 of cytarabine, forming a new prodrug, LA–Ara. The fatty acid chain with 12 carbons confers several advantages on the parent drug: (i) protecting the NH2 group from deactivation by deamination to an inactive metabolite; (ii) decreasing the introduction of inert material, leading to high drug loading (57.3% in theory); (iii) markedly raising the lipophilicity of Ara-C so that its cell and gastrointestinal membrane permeability were strikingly improved. Using a simple and common amidation reaction, the synthesis was successful, with a yield of 61.8%. The chemical constitution of the synthesized compound was characterized by 1H-NMR spectra, mass spectrometry (MS) and Fourier transform infrared spectroscopy (FTIR). The oil/water partition coefficient (lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) and the permeability of the cell membrane were determined to be 110 and 8.2 times those of the pure sample, respectively, indicating dramatically enhanced lipophilicity and penetrability. Unexpectedly, this unique amphiphilic molecule could self-assemble into nanofibers in the aqueous phase via a solvent displacement procedure. To assess the feasibility of the prodrug for oral administration, in vitro stability experiments were carried out. The cytotoxicity of the prodrug against HL60 and K562 cells was investigated, and the results revealed its superior antiproliferation efficiency over native Ara-C. Finally, the disposition of LA–Ara assembly after oral administration in vivo was studied on Wistar rats, and the results showed an outstandingly prolonged plasma half-life and higher oral bioavailability. In summary, this novel amphiphilic molecule improves the lipid solubility and anticancer activity of Ara-C, and the nanoassembly of LA–Ara may be a good platform to deliver Ara-C for oral medication applications.
P) and the permeability of the cell membrane were determined to be 110 and 8.2 times those of the pure sample, respectively, indicating dramatically enhanced lipophilicity and penetrability. Unexpectedly, this unique amphiphilic molecule could self-assemble into nanofibers in the aqueous phase via a solvent displacement procedure. To assess the feasibility of the prodrug for oral administration, in vitro stability experiments were carried out. The cytotoxicity of the prodrug against HL60 and K562 cells was investigated, and the results revealed its superior antiproliferation efficiency over native Ara-C. Finally, the disposition of LA–Ara assembly after oral administration in vivo was studied on Wistar rats, and the results showed an outstandingly prolonged plasma half-life and higher oral bioavailability. In summary, this novel amphiphilic molecule improves the lipid solubility and anticancer activity of Ara-C, and the nanoassembly of LA–Ara may be a good platform to deliver Ara-C for oral medication applications.
2. Materials
Cytarabine (Ara-C) was purchased from Aladdin Industrial Corporation. Lauric acid (LA) was purchased from J&K Chemical Ltd. Ethyl chloroformate (Et–O–CO–Cl) was purchased from Chengdu Beisite Reagent Co., Ltd. Sodium hydroxide, potassium dihydrogen phosphate and dipotassium hydrogen phosphate were purchased from Sinopharm Chemical Reagent Co., Ltd. Acyclovir was purchased from the National Institutes for Food and Drug Control, China. Double-distilled water was used in all experiments.3. Methods
3.1. Synthesis and purification of prodrug molecule LA–Ara
The prodrug was synthesized by linking the N4-amino group of cytarabine with lauric acid (Fig. 1) according to a previously described method.22,23 Briefly, lauric acid (0.1690 g, 0.845 mmol) was dissolved in anhydrous N,N-dimethylformamide (DMF, 5 mL). Triethylamine (0.0940 g, 0.930 mmol) and ethyl chloroformate (Et–O–CO–Cl) (0.1009 g, 0.930 mmol) in anhydrous DMF were added dropwise to the above solution under nitrogen. The reaction was maintained in an ice bath and stirred for 20 min. Then cytarabine (0.2466 g, 1.014 mmol), previously dissolved in anhydrous DMF, was added to the mixture and the reaction was allowed to react at room temperature for 3 days, during which the process was monitored by TLC. Finally, the mixture was concentrated to dryness under vacuum. The compound was washed with distilled water to remove impurities, and the lipophilic components were extracted with ethyl acetate. The organic phase was dried with anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by chromatography on SiO2, eluted with a gradient (from 80:1 to 30:1) of dichloromethane/methanol, to give the pure compound.
 | ||
| Fig. 1 The synthetic route of LA–Ara. | ||
3.2. Characterization of LA–Ara chemical structure
The chemical structure of LA–Ara was characterized by 1H-NMR (Bruker Avance 400 spectrometer) with DMSO-d6 as the solvent, electrospray tandem mass spectrometry (AB SCIEX API 4000) and Fourier transform infrared (FTIR) spectroscopy (Nicolet 6700).3.3. Saturation solubility and octanol/water partition coefficients (lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P)
P)
The saturation solubility of the new derivative of Ara-C in various media was determined by the thermostatic magnetic stirring method. Excess pure LA–Ara powder was dispersed in water and PBS buffer (pH 7.4) with or without 0.5% Tween-80, respectively. The suspensions were continuously stirred for 72 h at 100 rpm at 37.0 ± 1.0 °C. Then, 1 mL samples were removed and centrifuged at 14000 rpm for 10 min. The supernatants were diluted before high performance liquid chromatography (HPLC) analysis.
The octanol/water partition coefficients (lgP) of LA–Ara and Ara-C were determined by traditional shake-flask experiments. Briefly, 50 mL n-octanol and 100 mL distilled water in a capped Erlenmeyer flask were placed in an incubator shaker at 37.0 ± 1.0 °C. After equilibration for 24 h, the saturated oil and aqueous phases could be separated. LA–Ara dissolved in n-octanol (saturated with water) was added to an equal volume of saturated water in a plastic centrifuge tube. Conversely, Ara-C was dissolved in water (saturated with n-octanol) in the tubes, followed by the addition of saturated n-octanol for its water-solubility. These tubes were then stoppered and incubated at 37.0 ± 1.0 °C for 24 h. Finally, the oil phases were separated and analyzed by HPLC after centrifugation. lgP was calculated according to the following equation:
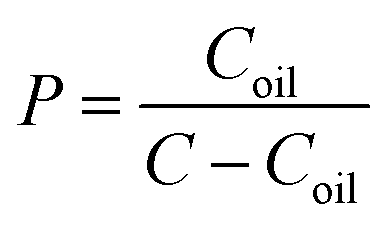
3.4. Preparation and TEM observation of LA–Ara nanoassembly
The LA–Ara nanoassembly was prepared by a nanoprecipitation method, in which the injection of a hydrophobic compound in a polar, water miscible solution into water (non-solvent) can generate a homogeneous dispersion of nanocolloids without the use of surfactant as a stabilizer.24 The nanoprecipitation method is especially suitable for preparing nanodispersions of drugs which are poorly soluble in water, such as our present system. 200 μL methanol containing 5 mg LA–Ara was added dropwise to stirring water under 800 rpm. After a while, the solution was centrifuged at 14000 rpm for 10 min and the sediment was redispersed with fresh water under ultrasonication. Finally, a suspension of LA–Ara nanoassembly was obtained. The morphology of the assembly was observed by transmission electron microscopy (TEM, JEM-200CX). The samples were prepared by dropping one drop of the suspension onto a copper grid before observation.
3.5. In vitro stability
000 rpm for 10 min. The supernatants were analyzed by HPLC.3.6. Cell culture
Human leukemic cell lines K562 (chronic granulocytic leukemic cell line) and HL60 (acute myeloblastic leukemic cell line) were kindly supplied by the Immunopharmacology Institute of Shandong University and the Shandong Analysis and Test Center. The two cell lines were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (GIBCO, Vitrogen Corporation), 100 units per mL penicillin and 100 mg mL−1 streptomycin, incubated in a humidified (37 °C, 5% CO2) incubator (SANYO), and grown in 30 mL culture flasks (Falcon BD).3.7. In vitro cytotoxicity
The cell inhibition of LA–Ara and Ara-C on the two leukemic cell lines were determined by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay. Exponentially growing K562 and HL60 cells were placed into 96-well plates at a density of 1 × 104 cells per 100 μL per well in media to which different concentrations of samples were added. After cultivation at 37 °C for 24 h and 48 h respectively, 10 μL of 5% MTT reagent (Dojindo, Kumamoto, Japan) was added to each well, followed by incubation for another 4 h. Then the medium was removed and the obtained blue formazan crystals were dissolved with DMSO. The optical density of each well was measured using a microplate reader (ELISA, Perkin Elmer) at a wavelength of 490 nm, and the cell inhibition rate of each sample was calculated according to the following equation:where Apositive control refers to the absorbance of cells without treatment, and Ablank refers to the absorbance of the culture medium without cells.

3.8. Parallel artificial membrane permeability assay (PAMPA)
Permeability studies were performed by the following protocol.25,26 A 96-well filter plate served as the receiving chamber, and a 96-well filter plate served as the donor compartment. 5 μL of an artificial membrane solution was uniformly coated on the hydrophobic filter material of a 96-well microtiter plate containing 1.5% (w/v) L-a-phosphatidylcholine (PC) and 0.5% (w/v) stearic acid (SA) in 1,7-octadiene. Immediately, 250 μL PBS (pH 7.4) containing 20 mM test compounds were added to the donor compartment, while the acceptor chamber was filled with 350 μL PBS (pH 7.4). The two plates were then coupled together and placed in a sealed container at 30 °C for 16 h. After incubation, samples from the receiver and donor wells were collected and analyzed by HPLC. The permeability of the compounds, Pam, was calculated according to the following equation:
where Vdn (mL) is the donor well volume, Vac (mL) is the acceptor well volume, S (cm2) is the membrane area (0.24 cm2), t (s) is the incubation time, Cdn (mM L−1) is the concentration of drug in the donor compartment, Cac (mM L−1) is the concentration of drug in the acceptor compartment, and Cequilibrium is the theoretical equilibrium concentration.

3.9. Pharmacokinetic studies in vivo
Healthy Wistar rats with body weight 205 ± 5 g were used in this study. All animal experiments were approved by the National Act on the use of experimental animals (People's Republic of China) and in full compliance with international ethics guidelines. After fasting for 12 h, the rats (n = 5 per treatment) were administered LA–Ara nanoassembly or Ara-C saline solution with equimolar doses of 15 mg kg−1 of cytarabine by gavage. At the predetermined time point, 0.5 mL blood samples were removed from the sinus jugular and placed into heparinized tubes that were prepared in advance. The plasma was immediately collected by centrifugation at 10000 rpm for 5 min and preserved at −20 °C until analysis.
Before determination, the plasma samples were prepared using a simple protein precipitation procedure. Briefly, 400 μL acetonitrile and 200 μL of internal standard (800 ng mL−1, acyclovir) were added to 200 μL of plasma, followed by vortexing for 1 min. After centrifugation, the supernatants were separated and evaluated by HPLC-MS/MS. The quantitative analysis was performed using a triple quadrupole mass spectrometer, Agilent 1260, equipped with an ESI (electrospray ionization) source, and a C18 column (150 mm × 4.6 mm i.d. 5 μm, Thermo) coupled with a Phenomenex C18 guard column (4 mm × 3.0 mm i.d. 5 μm). Gradient elution was applied to separate the analytes from endogenous materials using methanol and water containing 0.2% formic acid as the mobile phase at a flow rate of 0.60 mL min−1. The ESI source was set in positive ionization mode and transitions of 224 to 112 for Ara-C, 226 to 152 for acyclovir, and 426 to 294 for LA–Ara were used for quantification. The analysis methodology was established and the results are shown as Fig. S1–S4 in the ESI.†
3.10. Statistical analysis
Three replicates of each experiment were used, and the data were presented as mean ± standard deviation (SD). The statistical differences among groups were tested by Student's t-test (SPSS statistics 19.0), and a value of p < 0.05 was considered to be significant.4. Results and discussion
4.1. Synthesis and characterization of LA–Ara prodrug
The final product was obtained as a white powder in a yield of 61.8%. The chemical structure of synthesized LA–Ara was verified by 1H-NMR, as shown in Fig. S5,† and the data was as follows (DMSO-d6, d, ppm): 10.78 (s, 1H, –NH–), 8.05 (d, 1H, J = 7.60 Hz, 6 H), 7.20 (d, 1H, J = 7.60 Hz, 5 H), 6.06 (d, 1H, 1′-H), 5.47 (br, 2H, 2′-OH, 3′-OH), 5.06 (t, 1H, J = 11.2 Hz, 5′-OH), 4.03–4.10 (m, 1H, 2′-H), 3.90–3.96 (m, 1H, 3′-H), 3.80–3.87 (m, 1H, 4′-H), 3.61 (t, 2H, J = 14.8 Hz, 5′-H), 2.38 (t, 2H, a-H), 1.21–1.33 (m, 16H, b-H), 1.47–1.61 (m, 16H, c-H), 0.853 (t, 3H, J = 13.2 Hz, d-H). Overall, the appearance of an amide signal (10.78 ppm) confirmed the formation of a covalent bond between LA and Ara-C.Additionally, FTIR spectra and ESI-MS/MS spectra were acquired to confirm the chemical structure of LA–Ara. In Fig. S6,† obvious absorption bands for primary amines at 3476 and 3441 cm−1 were observed in the FTIR spectrum of Ara-C, while they were replaced by a broad absorption band of N–H stretching centered at 3348 cm−1 in the LA–Ara spectrum. Absorption peaks in the region of 2925–2853 cm−1 corresponding to methylene and methyl groups appeared in the spectra of both LA and the LA–Ara prodrug. In addition, an amide I absorption (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode) and an amide II absorption (mainly N–H bending and C–H stretching modes) were observed at 1704 cm−1, 1645 cm−1 and 1555 cm−1, respectively. All these findings corroborate the successful synthesis of LA–Ara. ESI-MS for C21H35O6N3 (M + H+) is 425.6 (Fig. S7†), which is consistent with the theoretically calculated value.
O stretching mode) and an amide II absorption (mainly N–H bending and C–H stretching modes) were observed at 1704 cm−1, 1645 cm−1 and 1555 cm−1, respectively. All these findings corroborate the successful synthesis of LA–Ara. ESI-MS for C21H35O6N3 (M + H+) is 425.6 (Fig. S7†), which is consistent with the theoretically calculated value.
4.2. Saturation solubility and octanol/water partition coefficients (lgP)
To investigate the physicochemical properties of the novel derivative of cytarabine, its saturation solubility in various media and its lgP were determined. As the results show in Table 1, the solubilities of LA–Ara in water and PBS (pH 7.4) were 10.13 μg mL−1 and 1.92 μg mL−1, respectively. Compared with our previous report of pure Ara-C,20 of which the solubility in water was 437.4 mg mL−1, the hydrophilicity of LA–Ara decreased substantially. The coefficient (P), a crucial physicochemical parameter to characterize the hydrophilicity and hydrophobicity of a drug, is always given as the logarithm to the base 10 (lgP).27 At 37 °C, the P value of LA–Ara was almost 110 times that of Ara-C, suggesting greater lipophilicity of LA–Ara compared to the pure drug. On the whole, the hydrophobic part (LA) of the new prodrug weakened the solubility of Ara-C and enhanced its lipophilicity, which can give rise to better absorption in vivo.28
P of LA–Ara and Ara-C
| A | |||
|---|---|---|---|
| Dissolution medium | Water | PBS (pH 7.4) | PBS (pH 7.4) with 0.5% Tween-80 |
| a p < 0.01. | |||
| Saturation solubility (μg mL−1) | 10.13 ± 0.06 | 1.92 ± 0.04 | 252.13 ± 0.10 |
| B | ||
|---|---|---|
| Pa | lgP |
|
| Ara-C | 0.04 ± 0.01 | −1.36 ± 0.13 |
| LA–Ara | 8.87 ± 0.38 | 0.95 ± 0.02 |
4.3. TEM observation of LA–Ara nanoassembly
The as-prepared LA–Ara nanoassembly in water appeared as a white flocculent suspension, which remained stable for more than two weeks under room temperature. The morphology of the assembly was observed by TEM, and the images are shown in Fig. 2. It can be seen that an abundance of nanofibers were formed by self-assembly of the amphiphilic LA–Ara molecule. These nanofibers with high aspect ratios curled and folded into slightly twisted ribbons approximately 30 nm in width. The unique formation of the nanostructure may be ascribed to H-bond formation between the amide groups in the cytarabine molecules. Meanwhile, hydrophobic interactions between the liposoluble tails with 12 carbons may also induce an ordered arrangement of the amphiphilic molecules, which facilitates the assembly of LA–Ara nanofibers.21,29,30 | ||
| Fig. 2 TEM images of LA–Ara nanofiber assembly at different magnifications (A and B). The scale bar of the inset is 200 nm. | ||
4.4. In vitro stability
In order to predict the stability of LA–Ara in vivo, studies were performed at 37 °C in PBS buffer with different pH values as well as in artificial gastrointestinal fluids; the results are shown in Fig. 3. It could be observed that the chemical hydrolysis of LA–Ara was a pH-dependent process, and the degradation rate increased as the pH decreased. In neutral conditions (pH 7.32), 98.04% of the intact LA–Ara molecule remained; however, only 81.61% and 67.53% of the original amount was left after incubation at pH 6.78, 4.51 and 1.21 for 12 h. In acidic environments, the degradation curves displayed a rapid decline in the first 2 h and leveled off afterward, especially at pH 1.21. On the basis of chemical structure, the amide bond of LA–Ara is susceptible to hydrolysis. Meanwhile, the hydrolysis rate of the amide bond is related to steric hindrance, i.e. the length of the spacer and cis–trans isomerism.31,32 Consequently, the initial sharp degradation of LA–Ara is presumably due to the minimal steric hindrance of the amide bond in the specially configured portion of the molecules, which exposes them to rapid cleavage. Generally, the human stomach is emptied every 2 h; 75.14% of intact LA–Ara remained after 2 h incubation at pH 1.21, which indicates that the stability of the prodrug would be influenced by the acidic conditions in the gastric environment. | ||
| Fig. 3 Chemical stability (A) and gastrointestinal stability (B) of LA–Ara prodrug investigated in different media. Each point represents the mean value of three independent experiments ± SD (error bars). | ||
The degradation kinetic curves of LA–Ara in artificial gastrointestinal fluids are shown in Fig. 3B. Within the first 2 h, elimination of the prodrug in artificial gastric juice was rapid, and 74.31% of LA–Ara remained at the end; this was similar to the results for LA–Ara incubated in the absence of enzymes (Fig. 5A, pH 1.21) for the same amount of time. However, the hydrolysis of the prodrug was negligible in artificial intestinal fluids for a further 6 h. It could be concluded that LA–Ara was easily hydrolyzed to the parent drug at low pH, while the catalytic effect of enzymes was inconspicuous, and that most parts of the prodrug could be stable in the gastrointestinal tract and would thus be absorbed into circulation. Namely, LA–Ara appears to be very appropriate for oral administration, which provides the theoretical foundation for pharmacokinetic studies in vivo.
It has been reported that self-assembly formulations are influenced by the pH values of their environment.33,34 The stability of the LA–Ara nanofibers was tested in different acidic media and recorded by TEM observation, as displayed in Fig. 4. At pH 1.2, the LA–Ara nanofibers broke into short fibers that crossed each other; few long nanofibers could be found after 2 h. When the incubation time was extended to 4 h, the short nanofibers were reduced and replaced by layered nanosheets. Meanwhile, at pH 4.5 and 6.8, the morphology of the nanofibers maintained the original formulation for 6 h, with little reduction in fiber length (Fig. S8†). The nanoassembly was prone to aggregation and shrinkage in acidic conditions.35 The cleavage of the long fibers at pH 1.2 may be ascribed to hydrolysis under acidic conditions, which disrupted the interaction between the inner H-bonds. However, the remaining fragments did not affect the oral administration.
 | ||
| Fig. 4 TEM images of LA–Ara nanofibers after incubation for 2 h (A, B and C) and 4 h (D, E and F) at pH 1.2, 4.5 and 6.8, respectively. | ||
The physiological stability of the LA–Ara assembly was investigated in simulated digestive juices, and the results are shown in Fig. S9.† After incubation for 2 h, the nanofiber assembly transformed into short fibers in artificial gastric juice (∼pH 1.2) (Fig. S9A†), while a large amount of nanofibers still existed after incubation for 4 h in artificial intestinal fluids (∼pH 7.5) (Fig. S9B†), indicating the physiological stability of the assembly in vitro.
4.5. In vitro cytotoxicity
MTT assays were applied to investigate the cytotoxicity of LA–Ara and Ara-C. Fig. 5 shows the inhibitory effects of the samples on the growth of HL60 cells and K562 cells after 24 and 48 h incubation. Obviously, the inhibitory rate of the two samples on the leukemia cells displayed dose- and time-dependencies. For HL60 cells after 24 h, the cytotoxicity of Ara-C was slightly higher than that of LA–Ara, with concentrations no greater than 60 μM. However, when the concentration was increased to 125 μM, the inhibition ratio of LA–Ara dramatically increased to 87.16% (a 2.33-fold increase), while just 5% growth was found for Ara-C. Furthermore, at the highest concentration (500 μM), the cell mortality was merely 58.11% for the parent drug compared with 96.38% of LA–Ara, implying significantly enhanced cytotoxicity for the prodrug. As the cultivation time was prolonged to 48 h, the inhibition ratio of the LA–Ara samples continued to increase, while hardly any increase in cell growth inhibition was found for Ara-C at each concentration. Overall, the LA–Ara molecules demonstrated a longer action time and higher cytotoxic activity compared with the pure drug. Similar properties were found for the inhibitory process of K562 cells. After 24 h incubation, the mortality rates of cells treated with Ara-C were considerably lower than those of LA–Ara for all doses (p < 0.01). The inhibition rate of LA–Ara already exceeded 50% at 125 μM and increased to 95.30% at the maximum concentration, while the mortality percentage of cells incubated with Ara-C was barely 28.78% under identical conditions. After 48 h incubation, a prominent decline in cell viability was observed in samples with both Ara-C and LA–Ara. As the dosage increased, the inhibition rate of Ara-C increased and the peak inhibitory ratio was less than 55%. By contrast, the inhibition rate of LA–Ara was already 58% at the lowest concentration, and the leukemic cells almost completely died under the maximal dose, suggesting that the prodrug exhibited stronger cytotoxicity on K562 cells. Broadly speaking, the cytotoxic ability of LA–Ara toward leukemia cells revealed a distinct concentration dependence and remarkable enhancement over the parent drug. This can probably be ascribed to the improved lipophilicity of Ara-C by coupling it with LA, leading to accelerated intracellular uptake for LA–Ara and thus improving the inhibition effect of the parent drug.36 During passive diffusion into cancer cells, a larger amount of drug had enhanced membrane permeability, which resulted in stronger cytotoxicity for LA–Ara. By comparison, the rate of penetration across the cellular membrane of Ara-C was limited by incubation time, due to the hydrophilic nature of the compound. Consequently, a longer reaction time induced a higher inhibition rate toward the tumor cells.37 | ||
| Fig. 5 Cell inhibition rates of different compounds toward K562 and HL60 cells after 24 h and 48 h incubation, **p < 0.01, n = 3. | ||
IC50 values (concentration resulting in 50% cell death) were calculated to quantitatively estimate the cytotoxic capability of the samples, and the data are presented in Table 2. It can be observed that the IC50 values of LA–Ara are distinctly lower than those of Ara-C for K562 cells at all incubation times, which is indicative of the superior anti-cancer activity of the prodrug. Although HL60 was more sensitive to Ara-C,20 the IC50 values of pure drug samples were still 4-fold higher than that of LA–Ara after both 24 and 48 h. On balance, LA–Ara rapidly kills leukemic cells and possesses stronger toxicity than Ara-C due to its modification, indicating that LA–Ara is a preferable therapy for leucocythemia.
| Samples | HL60 | K562 | ||
|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | |
| Ara-C | 211.62 | 205.72 | 60731.18 |
200.59 |
| LA–Ara | 54.40 | 46.15 | 92.23 | 16.84 |
4.6. Parallel artificial membrane permeability assay (PAMPA)
PAMPA is a preferred method to assess the rate and extent of membrane permeability. Herein, an artificial film solution simulating the alimentary canal was employed to examine the absorption of LA–Ara in vivo and the permeability coefficients (Pam) of the compounds; the results are shown in Table 3. For a reasonable comparison, the samples contained equal amounts of Ara-C. The data demonstrated that the Pam value of the native drug was remarkably increased by 8-fold of LA–Ara from the t-test result (p < 0.01). The enhanced permeability is attributed to the conjugation of the lipid moiety, LA, which improves the affinity with the membrane. Additionally, the modification of Ara-C shields the polar NH2 group and further increases the lipophilicity as well as the permeability. These findings suggested that the synthesized amphiphilic molecules would achieve better absorption in the gastrointestinal tract and higher oral bioavailability in contrast with the pure drug.4.7. Pharmacokinetic studies in vivo
To date, derivatives of Ara-C for oral application are attractive to researchers, and some designed derivatives have undergone phase 2 studies in patients.38,39 In the current study, nanoprecipitation was applied to prepare LA–Ara nanofibers. The use of organic solvent and the long length of the assembly restrict the intravenous administration of LA–Ara nanofibers. Overall, the as-prepared LA–Ara nanofibers in the present study are more suitable for oral administration. Pharmacokinetic performance studies were conducted to evaluate whether the synthesized prodrug strategy could improve the oral bioavailability of Ara-C. After oral administration of the LA–Ara assembly and Ara-C solution with equivalent amounts of effective drug in rats, the concentrations of Ara-C and its prodrug in the plasma samples were determined with a validated HPLC-MS/MS method. Plasma concentration–time profiles of the two samples are shown in Fig. 6. After feeding with pure Ara-C solution, the concentration of Ara-C increased to maximum at 1 h and rapidly decreased to almost zero within 12 h (Fig. 6, the insert curve a). Moreover, the drug was undetectable in plasma after 24 h, which indicated the low blood concentration and shorter action time of the unmodified drug with oral administration. In the case of the LA–Ara assembly, concentrations of both LA–Ara and Ara-C were detected in plasma samples. The drug–time curve of LA–Ara (Fig. 6b-1) was similar to that of Ara-C in the control group. The peak time was 1.5 h, and the prodrug could be monitored in vivo up to 36 h. Nevertheless, the concentration of Ara-C released from the prodrug achieved a much higher level in plasma (Fig. 6b-2). Additionally, the peak time was prolonged to 5 h and a large amount of Ara-C was retained in the blood circulation when the analysis was complete. These results supplied evidence that LA–Ara could be rapidly converted to the parent drug after oral administration and could noticeably increase the plasma concentration as well as the retention time of Ara-C in vivo compared with the unmodified drug. | ||
| Fig. 6 In vivo release of cytarabine in rats (n = 5) after administration of different formulations. (a) Cytarabine solution: detecting cytarabine. (b) LA–Ara assembly: detecting LA–Ara (b-1) and cytarabine (b-2). The inset is the magnification for a short time. | ||
The pharmacokinetics data were processed with DAS 2.1.1 (Drug And Statistics), a commonly used statistical software for new drug development, and the pharmacokinetic parameters are presented in Table 4. As can be observed from the results, the t1/2 and MRT0–∞ of LA–Ara were significantly longer than those of Ara-C, although there was little difference between their Cmax values. What is noteworthy is that the peak concentration of Ara-C from LA–Ara in the test group was 2773.65 ng mL−1, approximately 5-fold greater than that of pure Ara-C. Meanwhile, the clearance rate was much lower (0.03 times that of Ara-C). Last but not least, the total amount of LA–Ara (measured as Ara-C) represented by AUC0–∞ was 57579.54 ng h mL, with a 32.82-fold increase of pure Ara-C (AUC0–∞ = 17.5487 ng h mL).
| Parameters | Ara-C solution | LA–Ara assembly | |
|---|---|---|---|
| LA–Ara | Ara-C | ||
| a AUC0–∞ is the mean area under the concentration–time curve from time 0 to the last time point.b Cmax is the peak plasma concentration of the drug.c Tmax is the period of time to reach peak plasma concentration.d t1/2 is the elimination half-life of the drug.e MRT0–∞ is the mean residence time, describing the average time that drug molecules reside in the body.f Cl is the volume of plasma containing drug eliminated from the body per unit time, which represents the clearance rate of the drug from blood. | |||
| AUC0–∞/(ng h mL−1)a | 1754.87 | 10246.34 |
57579.54 |
| Cmax/(ng mL−1)b | 581.76 | 529.98 | 2773.65 |
| Tmax/(h)c | 1.00 | 1.50 | 5.00 |
| t1/2/hd | 1.80 | 39.73 | 11.19 |
| MRT0–∞/he | 3.62 | 42.58 | 17.42 |
| Cl/(L h−1)f | 8.55 | 2.56 | 0.261 |
The results clearly showed that the pharmacokinetic behavior of Ara-C changed greatly after oral administration of LA–Ara. As the prodrug hydrolyzed into Ara-C continuously in vivo, higher plasma concentrations and longer residence times were attained; also, the oral bioavailability of Ara-C was strikingly enhanced. After conjugation to the hydrophobic chain, the lipophilicity and ability to passively cross the lipid membrane were improved conspicuously, which was evidenced by the lgP and PAMPA assays. The compact configuration of the nanofibers provided a drug reservoir, realizing sustained drug release over a long period of time. Additionally, the freely circulating derivatives were resistant to deamination and inactivation due to the protected 4′-NH2 of Ara-C. All these were beneficial to the diffusion of most of the Ara-C into the circulatory system, resulting in an outstanding amelioration of its oral availability.
5. Conclusion
A new prodrug of cytarabine was designed by incorporation of LA; this prodrug could overcome drawbacks of the pure drug, such as low lipophilicity, short plasma half-life and rapid inactivation. lgP values and PAMPA results demonstrated that the LA–Ara exhibited better lipophipicity and permeability in comparison with Ara-C due to the fatty acid linkage. In aqueous media, the synthesized LA–Ara amphiphilic molecules could self-organize into nanofibers by a nanoprecipitation method with impressive drug loading. The cytotoxicity of LA–Ara was dramatically increased compared with Ara-C, and the IC50 values of the prodrug were only about 1/4 and 1/12 of that of Ara-C after 48 h incubation with HL60 and K562 cells, respectively. In vitro stability assays demonstrated that the prodrug was very suitable for oral administration. In vivo release studies showed that LA–Ara could be hydrolyzed to free cytarabine. Moreover, the accumulation and blood retention time of the released Ara-C were significantly improved, which indicated that the prodrug assembly would maintain a relatively high level of cytarabine for a long period of time. In conclusion, LA–Ara is a promising oral delivery system of Ara-C with a prominent therapy index for leukemia.
Acknowledgements
We gratefully acknowledge the financial support from National Natural Science Foundation of China (NSFC, No. 21373126).References
- M. G. Pallavicini, Pharmacol. Ther., 1984, 25, 207–238 CrossRef CAS PubMed.
- M. Rustum and R. A. Raymakers, Pharmacol. Ther., 1992, 56, 307–321 CrossRef PubMed.
- S. Grant, Adv. Cancer Res., 1997, 72, 197–233 CrossRef.
- J. H. Bryan, E. S. Henderson and B. G. Leventhal, Cancer, 1974, 33, 539–544 CrossRef CAS PubMed.
- J. J. Furth and S. Cohen, Cancer Res., 1968, 28, 2061–2067 CAS.
- R. L. Capizzi, J. C. White, B. L. Powell and F. Perrino, Semin. Hematol., 1991, 28, 54–69 CAS.
- G. D. Gray, J. A. Crim and M. M. Michelson, Transplantation, 1968, 6, 818–825 CrossRef CAS PubMed.
- A. F. Hadfield and A. C. Sartorelli, Adv. Pharmacol. Chemother., 1984, 20, 21–67 CAS.
- C. Luo, J. Sun, B. Sun and Z. He, Trends Pharmacol. Sci., 2014, 35, 12–22 CrossRef PubMed.
- S. Réjiba, L. H. Reddy, C. Bigand, C. Parmentier, P. Couvreur and A. Hajri, Nanomedicine, 2011, 7, 841–849 Search PubMed.
- X. Tao, J. Wang, Q. Feng, S. Gao, L. Zhang and Q. Zhang, Eur. J. Pharm. Biopharm., 2012, 82, 401–409 CrossRef CAS PubMed.
- Y. Jin, Y. Lian, L. Du, S. Wang, C. Su and C. Gao, Int. J. Pharm., 2012, 430, 276–281 CrossRef CAS PubMed.
- C. L. Neil, P. F. Wiley, R. C. Manak and T. E. Moxley, Cancer Res., 1970, 30, 1047–1054 Search PubMed.
- M. G. Sarpietro, S. Ottimo, M. C. Giuffrida, F. Rocco, M. Ceruti and F. Castelli, Int. J. Pharm., 2011, 406, 69–77 CrossRef CAS PubMed.
- B. S. Chhikara, D. Mandal and K. Parang, Eur. J. Med. Chem., 2010, 45, 4601–4608 CrossRef CAS PubMed.
- S. C. Tobias and R. F. Borch, Mol. Pharm., 2003, 1, 112–116 CrossRef.
- E. P. Cheon, J. H. Hong and H. K. Han, J. Pharm. Pharmacol., 2006, 58, 927–932 CrossRef CAS PubMed.
- E. P. Cheon and H. K. Han, Acta Pharmacol. Sin., 2007, 28, 268–272 CrossRef CAS PubMed.
- Y. Sun, J. Sun, S. Shi, Y. Jing, S. Yin, Y. Chen, G. Li, Y. Xu and Z. He, Mol. Pharm., 2008, 6, 315–325 CrossRef PubMed.
- F. Li, J. Liu, J. Shi and Y. Luan, RSC Adv., 2015, 5, 25382–25388 RSC.
- J. Liu, N. Ma, D. Zhao, Z. Li and Y. Luan, Colloids Surf., B, 2015, 136, 918–927 CrossRef CAS PubMed.
- L. Du, Y. Lian and Y. Jin, Colloids Surf., A, 2012, 393, 60–65 CrossRef.
- P. Couvreur, B. Stella, L. H. Reddy, H. Hillaireau, C. Dubernet, D. Desmaële, S. L. Mouelhi, F. Rocco, N. D. Bosquet, P. Clayette, V. Rosilio, V. Marsaud, J. M. Renoir and L. Cattel, Nano Lett., 2006, 6, 2544–2548 CrossRef CAS PubMed.
- L. Bildstein, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2011, 63, 3–23 CrossRef CAS PubMed.
- P. S. Hiremath, K. S. Soppimath and G. V. Betageri, Int. J. Pharm., 2009, 380, 96–104 CrossRef CAS PubMed.
- Z. S. Teksin, P. R. Seo and J. E. Polli, AAPS J., 2010, 12, 238–241 CrossRef CAS PubMed.
- A. Noubigh, A. Mgaidi and M. Abderrabba, J. Chem. Eng. Data, 2010, 55, 488–491 CrossRef CAS.
- W. J. Wechter, M. A. Johnson, C. M. Hall, D. T. Warner, A. E. Berger, A. H. Wenzel, D. T. Gish and G. L. Neil, J. Med. Chem., 1975, 18, 339–344 CrossRef CAS PubMed.
- L. C. Palmer, C. Y. Leung, S. Kewalramani, R. Kumthekar, C. J. Newcomb, M. O. Cruz, M. J. Bedzyk and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 14377–14380 CrossRef CAS PubMed.
- A. G. Cheetham, P. Zhang, Y. Lin, L. L. Lock and H. Cui, J. Am. Chem. Soc., 2013, 135, 2907–2910 CrossRef CAS PubMed.
- C. Mura, D. Valenti, C. Floris, R. Sanna, M. A. D. Luca, A. M. Fadda and G. Loy, Eur. J. Med. Chem., 2011, 46, 4142–4150 CrossRef CAS PubMed.
- M. D. Erion, K. R. Reddy, S. H. Boyer, M. C. Matelich, J. G. Galeno, R. H. Lemus, B. G. Ugarkar, T. J. Colby, J. Schanzer and P. D. Poelje, J. Am. Chem. Soc., 2004, 126, 5154–5163 CrossRef CAS PubMed.
- L. Qin, F. Xie, P. Duan and M. Liu, Chem.–Eur. J., 2014, 20, 15419–15425 CrossRef CAS PubMed.
- T. J. Moyer, J. A. Finbloom, F. Chen, D. J. Toft, V. L. Cryns and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 14746–14752 CrossRef CAS PubMed.
- H. Harde, A. K. Agrawal and S. Jain, Drug Delivery Transl. Res., 2015, 5, 498–510 CrossRef CAS PubMed.
- E. Lepeltier, C. Bourgaux, A. Maksimenko, F. Meneau, V. Rosilio, E. Sliwinski, F. Zouhiri, D. Desmaële and P. Couvreur, Langmuir, 2014, 30, 6348–6357 CrossRef CAS PubMed.
- J. Liu, Y. Jiang, Y. Cui, C. Xu, X. Ji and Y. Luan, Int. J. Pharm., 2014, 473, 560–571 CrossRef CAS PubMed.
- J. Braess, M. Freund, A. Hanauske, G. Heil, C. Kaufmann, W. Kern, M. Schussler, W. Hiddemann and E. Schleyer, Leukemia, 1998, 12, 1618–1626 CAS.
- E. Schleyer, J. Braess, B. Ramsauer, M. Unterhalt, C. Kaufmann, S. Wilde, M. Schussler and W. Hiddemann, Leukemia, 1995, 9, 1085–1090 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02051h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |