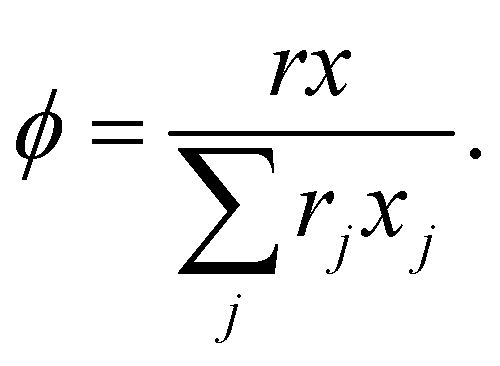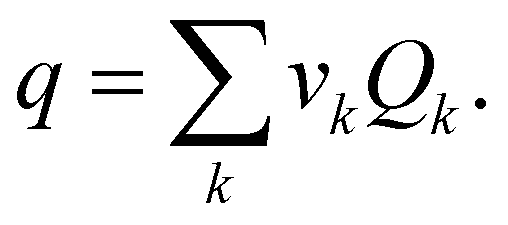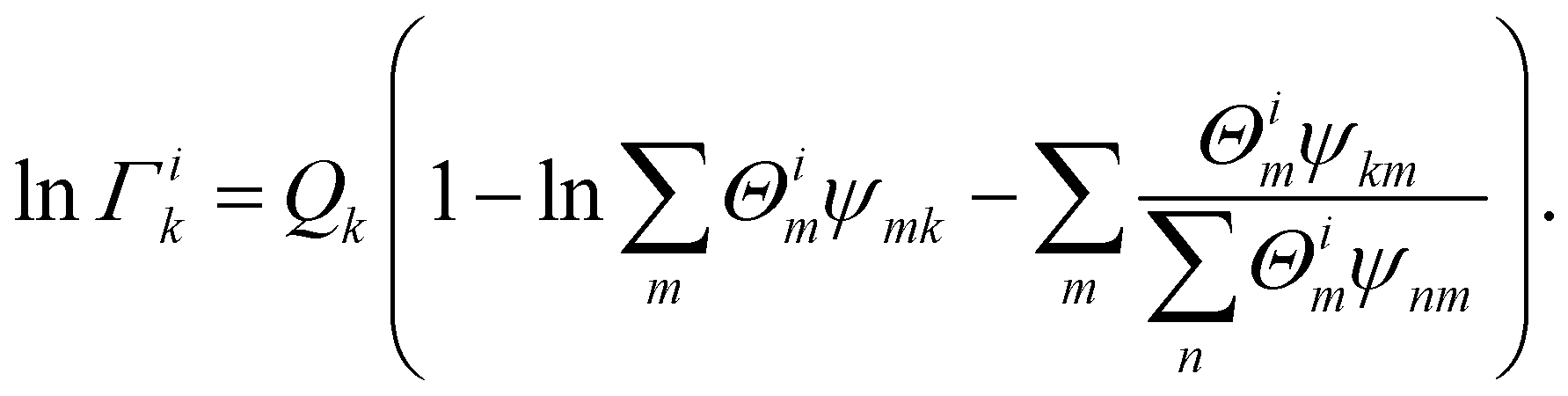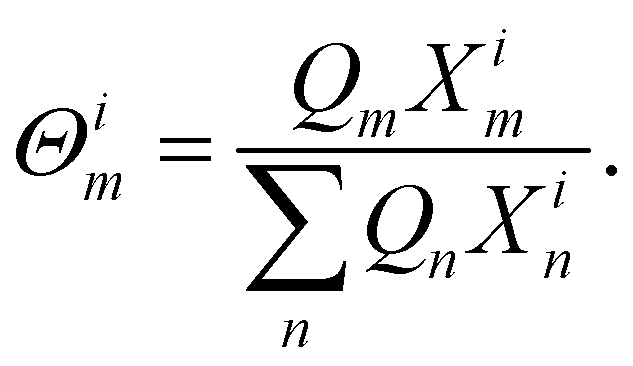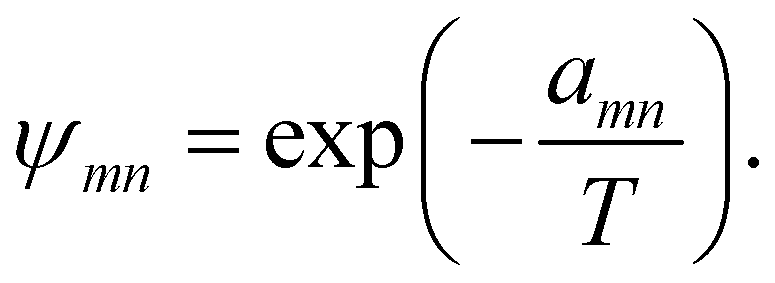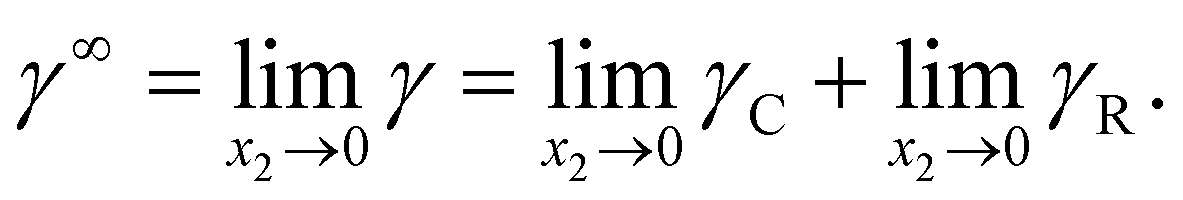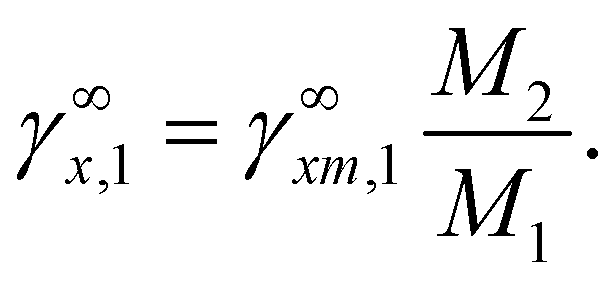DOI:
10.1039/C6RA02037B
(Paper)
RSC Adv., 2016,
6, 53643-53650
A modified UNIFAC-ZM model and phase equilibrium prediction of silicone polymers with ABE solution†
Received
23rd January 2016
, Accepted 13th May 2016
First published on 16th May 2016
Abstract
The UNIFAC model and its various modified models on behalf of the group contribution methods offer reliable knowledge of phase equilibrium data, which are making great contributions for separation processes. The application of the UNIFAC-ZM model for the silicone polymer system is not only restricted by the poor accuracy under a large temperature range, but also limited by the lack of SiO group related group interaction parameters. In this work, first, modification of the model was made with consideration of the temperature effect on group interactions. Then inverse gas chromatography (IGC), a simple method to determine the infinite dilution activity coefficient, was applied to determine the interaction parameters between the common groups CH3, OH, H2O, CH3CO and SiO contained in polydimethylsiloxane (PDMS) based on the equilibrium chromatography theory. The achieved model was further proved to agree with the experimental results well. The new model was also applied in the calculation of the partition equilibrium between acetone/butanol/ethanol water solutions of different concentrations and PDMS of different polymerization degrees and side chain length. All these results would not only help the improvement of UNIFAC model, but also instruct the separation processes of silicone polymer compounds.
1 Introduction
The behavior of small molecule penetrants for phase equilibrium is important in the design of many manufacturing and chemical engineering processes.1 For polymer membrane separation systems, confirmation of the solubility of components according to the activity is of great value for membrane material selection and mass transfer mechanism research.2 The most widely used polymer materials are silicone polymers which are distinguished by low volatility, low surface tension, low density, high compressibility and high resistance to thermal and photochemical degradation. These properties have led to the extensive technological applications of silicones, notably as membrane material. Polydimethylsiloxane (PDMS) of moderate selectivity and high permeability for many organics, is being widely applied in nano-filtration, reverse osmosis and pervaporation of organic-water separation such as biofuel acetone–butanol–ethanol (ABE) solution.3,4 Although a large number of phase equilibrium data for binary systems is available, much less data has been published for ternary-polymer systems. Because of the enormous time and high precision instrument needed for the measurement, thermodynamic theoretical model is especially required, which allows the calculation of the phase equilibrium behavior of multicomponent systems using only a limited number of experimental data, e.g., only binary data.
Group contribution method is just such a useful tool which treats the physical properties of pure material and mixture as the contribution sum of every group in the material molecule. The property of unknown material can be deduced from the relevance of group characteristic parameters with properties of known material.5 The first group contribution method for the prediction of vapor–liquid equilibria (VLE) was the ASOG (analytical solution of groups) method.6 Fredenslund et al.7 published the classical group contribution method Universal Quasichemical Functional Group Activity Coefficient model (UNIFAC) in 1975 based on the Universal Quasichemical Activity Coefficient model (UNIQUAC) and group analysis. The modifications of original UNIFAC model include correction of group volume parameter and surface area parameter, development of combinatorial term and energy correction with consideration of temperature.8–11 With the help of the different group contribution models, the various phase equilibria like VLE, liquid–liquid equilibria (LLE) and solid–liquid equilibria (SLE) as well as Gibbs energies, enthalpies of mixing and infinite dilution activity coefficients (γ∞) can be predicted.12–16 Quick screening of candidate solvents and separation processes (azeotropic or extractive distillation, extraction, absorption, solution crystallization and membrane separation) with phase equilibrium properties based on group contribution models can be very convenient in the early stages of a process design.17
Unfortunately, the group contribution methods discussed above also show the weak prediction for polymer system. Though models of UNIFAC-FV18,19 and Entropic-FV20 have been developed for polymer system and the precision is improved, the universality of free volume contribution is not enough and the usage is not convenient for the accurate volume parameter of polymer and small molecule. UNIFAC-ZM model proposed by Zhong et al.21 added a universal parameter for the modification of the combined term, which was different from other models. Besides, the group interaction parameters for polymer need to be supplemented for phase equilibrium.22 The group interaction parameters between the groups are mainly fitted to a great number of experimental equilibria data and stored in the Dortmund Data Bank.23 Because of the reliable results obtained and the large range of applicability, the different group contribution methods are used worldwide by a large number of chemical engineers.24 However, the equilibrium data of polymer and solvent is difficult to get for complex system or limited conditions, so it is important to achieve the group interaction parameters precisely with convenient experimental technology.25 Inverse gas chromatography (IGC) can determine the phase equilibrium of polymer at the infinite condition. IGC is a powerful method for study of the physico-chemical properties at infinite dilution with advantages of quick, convenience, accuracy and low consumption of reagent. Henry's constant, partition coefficient and mutual solubility are able to be achieved from the infinite dilution activity coefficients of vapor–liquid equilibria over the entire composition range.26–30 The missing group-contribution-based UNIFAC group interaction parameters were also able to be derived from infinite dilution activity coefficients.31–34
Thus in this study, optimization of UNIFAC-ZM model for predicting phase equilibrium of polymer systems was made with the consideration of temperature effect. The infinite activity coefficients of hexane, heptane, octane, acetone, butanol, ethanol and water with PDMS were determined by means of IGC in the temperature range of 373.2–413.2 K. SiO–CH3, SiO–CH3O, SiO–OH and SiO–H2O group interaction parameters were deduced via our corrected UNIFAC-ZM model which modified the temperature effect of group interaction. Then dissolution of ABE water binary solution in PDMS was reliably predicted and discussed with the model.
2 Modification of the UNIFAC-ZM model
The activity coefficient γ (mole ratio) of component in the mixture was built up by two terms according to the eqn (1),Where γC was the combinatorial term and γR the residual term. γC showed the contribution of molecule size and shape. In original UNIFAC model, the following combinatorial part8 was applied as eqn (2),| |
 | (2) |
where Z was the coordination number of component equaled 10. The volume fraction ϕ and surface area fraction θ was| |
 | (3) |
| |
 | (4) |
in which x was the mole fraction of the pure component. Based on van der Waals group volume and surface area, the volume parameter of group r and surface area parameter of group q could be achieved by eqn (5) and (6),| |
 | (5) |
| |
 | (6) |
where νk was the number of group k in the certain molecule j. Rk and Qk were the corrected van der Waals volume and surface area of the structural groups, respectively.35
For polymer solution, the molecule size effect was different from small molecule because of polymer chain effect. The excluded volume of monomer, dimer and trimer with overlapping hard-spheres was evaluated from geometrical consideration using the formula derived by Lustig et al.36,37 as follows,
Then the excluded volume of an n-mer chain molecule, Vn, is approximately
| | |
Vn ≈ V2 + (n − 2)(V3 − V2) = 2.75747nδ3 + 1.55364δ3.
| (10) |
For a long-chain polymer, the second term on the right of eqn (10) can be neglected
| | |
Vn ≈ 2.75747nδ3 = 0.6583nV1.
| (11) |
So the correction of γC of UNIFAC model was made as eqn (12) by Zhong et al.,21
| |
 | (12) |
in which
ϕ′
| |
 | (13) |
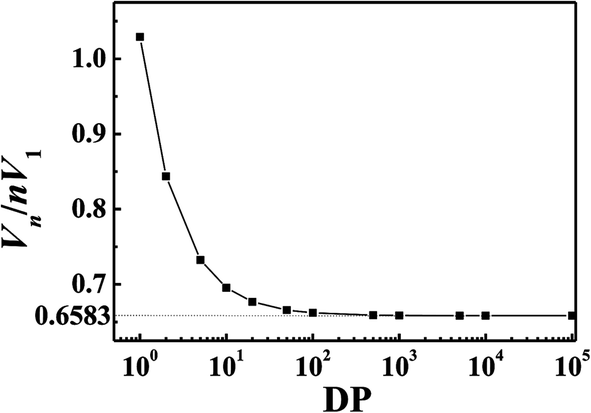
For small molecule, the polymer correction parameter ε equaled 1. For polymer, ε equaled 0.6583n. The application scope of UNIFAC-ZM model depended on the relative size of 2.75747n with 1.55364 as shown in Fig. 1. For most polymers, polymer degree (DP) was usually much larger than 10, which meant that the correction parameter 0.6583n was suitable for polymer or large molecule with repeat unit over 10.
 |
| | Fig. 1 Vn/nV1 changing with polymer degree. | |
The most acceptable UNIFAC model in recent years was modified UNIFAC model proposed by Gmehling et al.10 Their defined ϕ′ was
| |
 | (14) |
The index 3/4 was regressed from a great amount of equilibrium data. It was believable to apply ε for volume fraction correction with UNIFAC-ZM model instead of explicit term accounting for the volume change of polymer molecule.
Residual term γR was as eqn (15), which characterized the effect of intramolecular or intermolecular interaction and diffusion force as well as hydrogen bond.
| |
 | (15) |
In eqn (15), Γk was the residual activity of group k and Γpk was residual activity of group k referred to solution of pure molecule i for normalization. When xi approached 1, γi came near to 1. Γk and Γpk were calculated by eqn (16) and (17)
| |
 | (16) |
| |
 | (17) |
in which
m and
n were the number of group.
Θm and
Θim were the surface area fraction of group
k in total solution and certain molecule
i calculated by
eqn (18) and
(19)| |
 | (18) |
| |
 | (19) |
where
Xm was the mole fraction of group
m in the solution and
Xim the mole fraction of group
m in certain molecule
i.
Xm and
Xim was calculated by
eqn (20) and
(21)| |
 | (20) |
| |
 | (21) |
where
vim was the number of group
m in component
i and
xi mole fraction of component.
ψmn was the group interaction term, which could be calculated by eqn (22)
| |
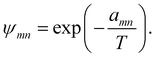 | (22) |
in which
amn was the (binary) group interaction parameter as the measure of interaction energy. The temperature-dependent group interaction parameters were introduced to eliminate the weakness of original UNIFAC-ZM model as modified UNIFAC model did in our work. The Modification of UNIFAC-ZM model was made as
eqn (23)| |
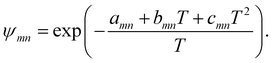 | (23) |
The SiO related group interaction parameters for the modified UNIFAC-ZM model were determined from the infinite dilution activity coefficients in the following part.
3 Determination of infinite dilute activity coefficient by IGC
Infinite dilution activity coefficient (γ∞) characterizes the behavior of a single solute molecule completely surrounded by solvent, which generally provides incisive information regarding solute–solvent interactions in the absence of solute–solute interaction.38 It offers a wider applicability than any measurement at finite concentration including prediction of the phase behavior of a mixture over the entire concentration range from the industrial viewpoint.39 IGC was the classical method for determination of γ∞, the principle of which was based on the equilibrium partitioning of probe molecules between vapor and stationary phase.
The generalized equation for γ∞ at the infinite condition was as eqn (24)40
| |
 | (24) |
where
R was the gas constant,
B11 the second term of the Virial EOS given by
| |
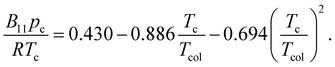 | (25) |
and
Vmol the molar volume of the solute. The saturated vapor pressure of the solvents
psat at different temperature was calculated from Antoine equation.
41 The specific retention volume of the solvent
Vsr was quantitatively determined as
eqn (26)42,43| |
 | (26) |
where
Qg was the volume flow rate, retention time
tr, dead time
t0,
ms the mass of polymer coated on the support material packed in the column, and
To the temperature of the flow meter. Since the gas was compressible, the pressure drop along the column might cause an increase of
Qg in the pressure of the outlet (
po) compared with the inlet value (
pi). Then correction factor
j was usually added for the correction of the
Qg as
eqn (27).
| |
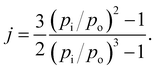 | (27) |
3.1 Inverse gas chromatography experiments
3.1.1 Materials. PDMS with viscosity of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cp and average molecular weight of 80000 was achieved by Beijing Second Chemistry Company of China. Hexane, heptane, octane, acetone, butanol, and ethanol of analytical grade were purchased from China Medicine Group (Shanghai Chemical Reagent Corporation). All the reagents and double distilled water were used without further purification.
000 cp and average molecular weight of 80000 was achieved by Beijing Second Chemistry Company of China. Hexane, heptane, octane, acetone, butanol, and ethanol of analytical grade were purchased from China Medicine Group (Shanghai Chemical Reagent Corporation). All the reagents and double distilled water were used without further purification.
3.1.2 Preparation of chromatographic column. The support was carefully poured into the PDMS–heptane solution. The heptane was steadily and fully volatized for at least 48 h at 333 K. The coated stationary phase was filled into the stainless steel column carefully with another end connected to a vacuum pump (0.1 Mpa) through a safety bottle. In this process, the column should be knocked continuously to disperse the support uniformly.
3.1.3 Chromatographic measurement method. Chromatographic measurements were carried out by GC-14C (Shimadzu Co. Ltd, Japan) equipped with the thermal conductivity detector (TCD). The flow vapor phase was solvent carried by inert gas hydrogen and the stationary phase was polymer membrane coating on the inert particles. Small volume of solvent was injected into the column oven as the disturbance like the infinite dilution via the syringe. Then, the output signal from the detector was fed back to the computer with chromatographic workstation for further analysis.The small pulse of solvent caused the partition of solvent in the polymer and the gas. The retention time tr of different solutes was recorded. The dead time t0 was measured by the alkane series (hexane–heptane–octane) method44 with the consideration of nitrogen retention in PDMS. Qg was determined by soap bubble flow meter and adjusted by the pressure drop. All the experiments were carried out for three times to reduce deviation.
3.2 Calculation of infinite dilute activity coefficient
γ∞ of hexane, heptane, octane (alkanes), acetone (A), butanol (B), ethanol (E) and water (W) were calculated according to the eqn (24). The γ∞ changing with temperature was drawn in Fig. 2.
 |
| | Fig. 2 γ∞ of solvents in PDMS. | |
Fig. 2(a) showed that γ∞ values of alkanes decreased with the increasing of temperature and carbon chain from 373.2 K to 413.2 K. Fig. 2(b) illustrated that γ∞ values of ABEW was in the order of acetone < butanol < ethanol < water. The difference of γ∞ also decreased at high temperature. γ∞ reflected the intermolecular interaction between the solvent and polymer and gave an idea of the polymer–solvent compatibility.45,46 The lower γ∞ meant the higher solubility. γ∞ of water was the highest obviously in those solutes due to the PDMS being a hydrophobic polymer. Solubility was elevated with increasing temperature, because the high-temperature caused the augment of mobility of the PDMS chains and the weakeness of the repulsive force between the molecules.
4 Fitting of group interaction parameter
For a binary system, the infinite dilution state was as follows: the peculiar situation of solution, as the concentration of one component approaches zero, while the mole fraction of the other component went to unity. In such a case, the activity coefficient of component was defined as the infinite dilution activity coefficients as eqn (28)| |
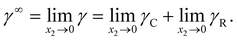 | (28) |
Then the group interaction parameter could be regressed by minimization of the following objective function using the Simplex–Nelder–Mead method as eqn (29)47
| |
 | (29) |
where
γ∞exp and
γ∞cal were the IGC experimental value and UNIFAC calculated value of
γ∞, respectively. The mass ratio and mole ratio infinite dilution coefficient were transformed by
eqn (30).
48| |
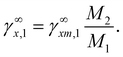 | (30) |
Rk and Qk of SiO were regressed together with group interaction parameters as modified UNIFAC model did.10 Some of the known parameters of modified UNIFAC model were applied in our modified UNIFAC-ZM model.49 All the γ∞exp and γ∞cal of original UNIFAC model, original UNIFAC-ZM model, modified UNIFAC model and modified UNIFAC-ZM model for hexane–PDMS, heptane–PDMS and octane–PDMS were shown in Fig. 3.
 |
| | Fig. 3 γ∞exp and γ∞cal of original UNIFAC model (O), original UNIFAC-ZM model (ZM), modified UNIFAC model (MO) and modified UNIFAC-ZM model (MZM). | |
As seen from Fig. 3, UNIFAC and UNIFAC-ZM model showed poor prediction ability of γ∞, which might be accused to the ignorance of temperature effect. The introduction of temperature-dependent interaction parameters for modified UNIFAC model and modified UNIFAC-ZM model permitted a more reliable description of the real phase behavior as a function of temperature. The average absolute deviations (AADs) for the γ∞ of alkanes were listed in the Table 1.
Table 1 Comparison of original UNIFAC model (O), original UNIFAC-ZM model (ZM), modified UNIFAC model (MO) and modified UNIFAC-ZM model (MZM) for γ∞ prediction
| System |
AADa/% |
| O |
ZM |
MO |
MZM |
 |
| Hexane |
33.0 |
36.0 |
19.9 |
4.74 |
| Heptane |
37.8 |
40.7 |
3.67 |
2.10 |
| Octane |
41.2 |
44.3 |
19.9 |
4.70 |
Table 1 showed that the original UNIFAC and UNIFAC-ZM model were not applicable and the modified UNIFAC-ZM model was better than the modified UNIFAC model for γ∞ of alkane–PDMS. The calculated Rk and Qk of SiO was 2.577 and 1.908. The group interaction parameters of SiO–CH3 for the modified UNIFAC-ZM model were listed in Table 2.
Table 2 SiO (Group n) related group interaction parameter for modified UNIFAC-ZM model
| Group m |
amn |
bmn |
cmn |
anm |
bnm |
cnm |
| CH3 |
9.802 × 102 |
−3.433 × 10−3 |
−6.646 × 10−3 |
4.239 × 102 |
−1.976 × 10−2 |
4.938 × 10−3 |
| CH3O |
−1.066 × 103 |
−1.395 × 10−2 |
1.149 × 10−2 |
4.324 × 103 |
−1.054 × 10−1 |
−2.009 × 10−2 |
| OH |
4.264 × 103 |
−1.135 × 10−1 |
−2.215 × 10−2 |
7.095 × 102 |
6.034 × 10−2 |
4.404 × 10−3 |
| H2O |
2.163 × 102 |
8.657 × 10−3 |
6.469 × 10−3 |
6.452 × 102 |
−3.890 × 10−3 |
−5.471 × 10−3 |
Based on the achieved Rk and Qk of SiO and SiO–CH3 group interaction parameters, other SiO–CH3O, SiO–OH and SiO–H2O for the modified UNIFAC-ZM model were regressed as eqn (29) and listed in Table 2. The achieved SiO related group interaction parameters were also applicable for the modified UNIFAC model. The γ∞exp and γ∞cal of modified UNIFAC-ZM model for acetone–PDMS, butanol–PDMS and ethanol–PDMS and water–PDMS were shown in Fig. 4.
 |
| | Fig. 4 γ∞exp and γ∞cal of modified UNIFAC-ZM model for ABEW with PDMS. | |
As can be seen from Fig. 4, the modified UNIFAC-ZM model showed good agreement with experimental results.
5 Prediction of solubility separation factor by modified UNIFAC-ZM model
Based on the optimization of UNIFAC-ZM model and SiO related parameters, it is convenient and flexible to predict phase equilibrium of various compounds from limited groups. Through the equal activity of component in solution and PDMS, the concentration of phase equilibrium in PDMS membrane wm1 and wm2 could be achieved for given concentration of solution wf1 and wf2.
The solubility separation factor of permselective component with subscript 2. Ss was defined as eqn (31), which was very important for PDMS membrane separation process.
| |
 | (31) |
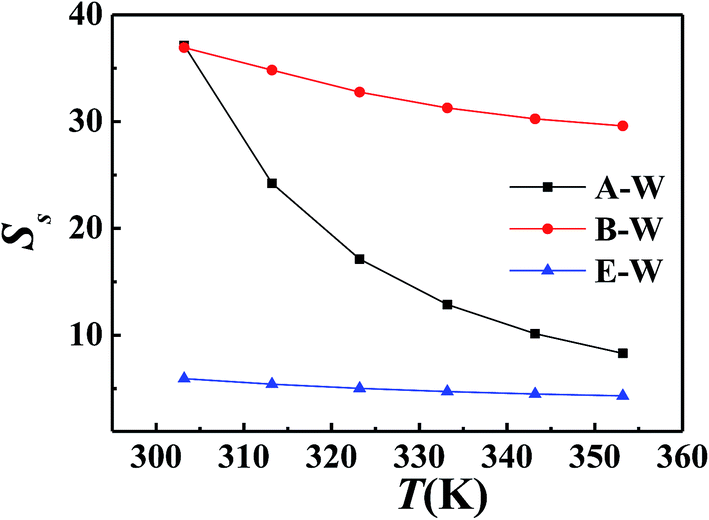
The effect of solution types and concentration, temperature, degree of polymerization and side chain length of PDMS on solubility separation factor were shown in Fig. 5–8 respectively.
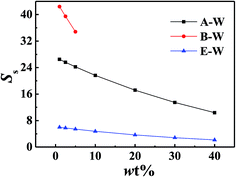 |
| | Fig. 5 Solubility separation factor of ABE–water binary solution with PDMS (Mn = 80000) at 313.2 K. | |
Fig. 5 showed PDMS had the best of butanol among A–E, B–E and W–E solution which implied the possibility of permselective of butanol with PDMS membrane for ABE fermentation. All the Ss decreased with the increasing of feed concentration. Phase separation occurred when butanol–water solution was over 10 wt%, which was proved by calculated activity of butanol over 1. So the phase equilibrium of higher concentration was not calculated. Fig. 6 showed all the Ss of the same concentration decreased with increasing temperature. Fig. 7 illustrated solubility selectivity decreased with increasing of polymerization degree when DP < 300. The reason might be the hydrophily of SiO group increased faster than CH3 group hydrophobicity. The even larger molecule weight made little contribution with Ss. Fig. 8 illustrated addition of hydrophobicity carbon side-chain length helped the improvement of solubility selectivity. Those conclusions needs to be further proved in our phase equilibrium experiment, the paper of which is in preparing.
 |
| | Fig. 6 Solubility separation factor of 5 wt% ABE–water binary solution with PDMS (Mn = 80000). | |
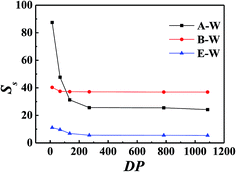 |
| | Fig. 7 Solubility separation factor of 5 wt% ABE–water binary solution with different polymerization degree (DP) of PDMS at 313.2 K. | |
 |
| | Fig. 8 Solubility separation factor of 5 wt% ABE–water binary solution with different length of side-chain at 313.2 K. | |
6 Conclusions
Modified UNIFAC-ZM model using an improved combinatorial part and temperature-dependent group interaction parameters was proposed in our work, which had good prediction ability. Through the determination of infinite dilute coefficient by IGC method in this report, the achieved group interaction parameter of SiO with CH3, OH, H2O, CH3O also supplemented the list of group interaction parameters for the modified UNIFAC model. The corrected model was tried in the phase equilibrium prediction of multicomponent polymer systems, the discussions of which might instruct the design and control of PDMS membrane separation process.
Phase equilibrium data is of paramount importance for processes involving separation of chemical species, while their reliable experimental determination is often difficult and time-consuming. The ability of reliably predicting the phase equilibrium behavior of binary and multicomponent systems opens a wide field of applications of industrial interest. Whereas the reliability of the results mainly depends on the quality of the group interaction parameters, the range of applicability depends on the size of the parameter matrix. It means that the parameters in this experiment could be further improved with larger phase experiments. The group interaction parameters might be defined according to the intrinsic physics properties of atoms from present fitted values in future.
Acknowledgements
We highly appreciated the financial supports of Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), National Natural Science Foundation of China (21576150), Science Foundation of Tsinghua University (20131089399) and the Special funds for technological development research of Research Institutes from National Ministry of Science and Technology (2013EG111129).
References
- J. Gmehling, D. Constantinescu and B. Schmid, Annu. Rev. Chem. Biomol. Eng., 2015, 6, 267–292 CrossRef CAS PubMed.
- L. P. Cunico, R. Ceriani, B. Sarup, J. P. O Connell and R. Gani, Fluid Phase Equilib., 2014, 362, 318–327 CrossRef CAS.
- G. Cocchi, M. G. De Angelis and F. Doghieri, J. Membr. Sci., 2015, 492, 612–619 CrossRef CAS.
- G. Cocchi, M. G. De Angelis and F. Doghieri, J. Membr. Sci., 2015, 492, 600–611 CrossRef CAS.
- A. Randová, L. Bartovská, S. Hovorka, M. Poloncarzová, Z. Kolská and P. Izák, J. Appl. Polym. Sci., 2009, 111, 1745–1750 CrossRef.
- E. L. Derr and C. H. Deal, Adv. Chem., 1973, 11–30 CAS.
- A. Fredenslund, R. L. Jones and J. M. Prausnitz, AIChE J., 1975, 21, 1086–1099 CrossRef CAS.
- I. Kikic, P. Alessi, P. Rasmussen and A. Fredenslund, Can. J. Chem. Eng., 1980, 58, 253–258 CrossRef CAS.
- E. R. Thomas and C. A. Eckert, Ind. Eng. Chem. Process Des. Dev., 1984, 23, 194–209 CAS.
- U. Weidlich and J. Gmehling, Ind. Eng. Chem. Res., 1987, 26, 1372–1381 CrossRef CAS.
- B. L. Larsen, P. Rasmussen and A. Fredenslund, Ind. Eng. Chem. Res., 1987, 26, 2274–2286 CrossRef CAS.
- D. C. Kannan, J. L. Duda and R. P. Danner, Fluid Phase Equilib., 2005, 228, 321–328 CrossRef.
- B. Khalfaoui and D. Newsham, J. Chromatogr. A, 1994, 688, 117–123 CrossRef CAS.
- H. P. Jung, E. L. Jung and P. W. Carr, J. Solution Chem., 1991, 20, 1189–1198 CrossRef.
- Y. Demirel, Can. J. Chem. Eng., 1990, 68, 697–701 CrossRef CAS.
- J. C. Bastos, M. E. Soares and A. G. Medina, Ind. Eng. Chem. Res., 1988, 27, 1269–1277 CrossRef CAS.
- J. W. Kang, V. Diky, R. D. Chirico, J. W. Magee, C. D. Muzny, I. Abdulagatov, A. F. Kazakov and M. Frenkel, Fluid Phase Equilib., 2011, 309, 68–75 CrossRef CAS.
- A. Fredenslund, Fluid Phase Equilib., 1989, 52, 135–150 CrossRef CAS.
- C. M. Tseng, M. S. Elaasser and J. W. Vanderhoff, J. Appl. Polym. Sci., 1986, 32, 5007–5019 CrossRef CAS.
- C. Wibawa, S. Takishima, Y. Sato and H. Masuoka, J. Appl. Polym. Sci., 2005, 97, 1145–1153 CrossRef.
- C. Zhong, Y. Sato, H. Masuoka and X. Chen, Fluid Phase Equilib., 1996, 123, 97–106 CrossRef CAS.
- J. Gmehling, P. Rasmussen and A. Fredenslund, Ind. Eng. Chem. Process Des. Dev., 1982, 21, 118–127 CAS.
- Dortmund data bank. Home page. http://www.ddbst.com.
- R. Wittig, J. Lohmann, R. Joh, S. Horstmann and J. Gmehling, Ind. Eng. Chem. Res., 2001, 40, 5831–5838 CrossRef CAS.
- W. D. Cai, N. Ramesh, F. Tihminlioglu, R. P. Danner, J. L. Duda and A. de Haan, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1046–1055 CrossRef CAS.
- S. T. Lin and S. I. Sandler, AIChE J., 1999, 45, 2606–2618 CrossRef CAS.
- M. Bader and K. Gasem, Can. J. Chem. Eng., 1998, 76, 94–103 CrossRef CAS.
- S. J. Zhang, T. Hiaki and K. Kojima, Fluid Phase Equilib., 2002, 198, 15–27 CrossRef CAS.
- E. C. Voutsas and D. P. Tassios, Ind. Eng. Chem. Res., 1997, 36, 4965–4972 CrossRef CAS.
- S. R. Sherman, D. B. Trampe, D. M. Bush, M. Schiller, C. A. Eckert, A. J. Dallas, J. J. Li and P. W. Carr, Ind. Eng. Chem. Res., 1996, 35, 1044–1058 CrossRef CAS.
- I. M. Balashova, R. G. Buduen and R. P. Danner, Fluid Phase Equilib., 2012, 334, 10–14 CrossRef CAS.
- B. Khalfaoui and D. Newsham, Fluid Phase Equilib., 1994, 98, 213–223 CrossRef CAS.
- S. M. Ashraf, M. Ramakrishna, D. Prasad and M. B. Rao, Chem. Eng. J., 1999, 75, 87–92 CrossRef CAS.
- H. Grensemann and J. Gmehling, Ind. Eng. Chem. Res., 2005, 44, 1610–1624 CrossRef CAS.
- D. S. Abrams and J. M. Prausnitz, AIChE J., 1975, 21, 116–128 CrossRef CAS.
- R. Lustig, Mol. Phys., 1986, 59, 195–207 CrossRef CAS.
- K. G. Honnell and C. K. Hall, J. Chem. Phys., 1989, 90, 1841–1855 CrossRef CAS.
- K. Kojima, S. Zhang and T. Hiaki, Fluid Phase Equilib., 1997, 131, 145–179 CrossRef CAS.
- J. Wisniak, H. Segura and R. Reich, Phys. Chem. Liq., 1996, 32, 1–24 CrossRef CAS.
- C. L. Young, Chromatogr. Rev., 1968, 10, 129–158 CrossRef CAS PubMed.
- I. H. Romdhane and R. P. Danner, AIChE J., 1993, 39, 625–635 CrossRef CAS.
- C. W. Zhao, J. D. Li, Z. Jiang and C. X. Chen, Eur. Polym. J., 2006, 42, 615–624 CrossRef CAS.
- J. M. Braun and J. E. Guillet, Macromolecules, 1975, 8, 882–888 CrossRef CAS.
- N. S. Wu, G. S. Wu and M. Y. Wu, J. Chromatogr. Sci., 2006, 44, 244–246 CAS.
- Q. G. Zhang, Q. L. Liu, J. Lin, J. H. Chen and A. M. Zhu, J. Mater. Chem., 2007, 17, 4889–4895 RSC.
- D. Patterson, Y. B. Tewari, H. P. Schreiber and J. E. Guillet, Macromolecules, 1971, 4, 356–359 CrossRef.
- J. Huang, J. Li, J. Chen, X. Zhan and C. Chen, Can. J. Chem. Eng., 2009, 87, 547–553 CrossRef CAS.
- S. L. Bacon, J. S. Parent and A. J. Daugulis, J. Chem. Technol. Biotechnol., 2014, 89, 948–956 CrossRef CAS.
- J. Gmehling, J. D. Li and M. Schiller, Ind. Eng. Chem. Res., 1993, 32, 178–193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02037b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.