DOI:
10.1039/C6RA01959E
(Paper)
RSC Adv., 2016,
6, 34625-34632
Hydroxyalkylation of phenol to bisphenol F over Al-pillared clay
Received
22nd January 2016
, Accepted 30th March 2016
First published on 31st March 2016
Abstract
Hydroxyalkylation of phenol to bisphenol F over the intercalation of aluminum hydroxy oligomeric into layered montmorillonite K10 was investigated. A remarkably high product yield (89.2%) and selectivity to bisphenol F (92.7%) has been achieved at a 110 °C reaction temperature and reaction time of 80 min with a Al-MMT(6) catalyst. A series of catalysts were prepared and characterized by FT-IR, XRD, BET, NH3-TPD and Py-IR. Characterization results showed that the catalytic performance of these catalysts depended on weak and moderate acidity and the textural properties (specific surface areas). The effect of the catalyst calcination temperature to this reaction was also studied. Moreover, the influences of various reaction parameters like mole ratio, catalyst concentration, reaction temperature and reaction time on the product yield and selectivity to bisphenol F were investigated. Finally, the reusability of the catalyst was studied and a plausible mechanistic pathway was proposed.
1 Introduction
The synthesis of bisphenol F has been given extensive attention due to the increasing demand and applicability in plastics, resins and rubber industries.1–3 Conventionally, hydroxyalkylation of phenol with formaldehyde to bisphenol F could be catalyzed by mineral acids, such as phosphoric acid, hydrochloric acid, sulfuric acid or other inorganic acids.4–6 Even though promising results have been attained, these homogeneous catalysts suffer from serious drawbacks such as hazards of handling, and corrosion of equipment. In addition, the recovery of homogeneous catalysts is an extremely formidable obstacle.7 Our group are highly desirable for solid catalysts to exhibit activities and selectivity at least comparable to homogeneous catalysts for bisphenol F synthesis. Solid acid catalysts that are insoluble, easily recovery, reuse, and highly active for eco-friendly chemical processes. A series of studies have been reported about the design of solid acid catalysts for bisphenol F synthesis, which spans over a broad range of catalytic materials including modified mesoporous silicas,8 clay,9 organometallic framework,10 and zeolites.11 Additionally, the PWA/MCM-41 was reported to be active for bisphenol F synthesis.12 The studies indicate that the concentration of acidic sites was a more predominant factor than its surface area for catalytic activity. The use of the PTA@MIL-101 (Fe or Cr) was also reported as efficient solid catalysts to produce bisphenol F by Chen et al.10 and leading to different bisphenol F isomer distributions. However, the synthesis of these materials is complex and need complicated equipment. In other work, one can underline the special affection in acid site for bisphenol F isomer distributions. Specifically, DTP/montmorillonite K10 were tested by Garade et al.,13 leading to different bisphenol F isomer distributions. However, the low catalytic activity severely restricted its application in the reaction that hydroxyalkylation of phenol with formaldehyde to bisphenol F.
In this work, we describe the catalyst prepared that Al-pillared clays via aluminum hydroxy oligomeric intercalated in montmorillonite K10, and evaluated for the hydroxyalkylation of phenol to bisphenol F. Furthermore, the effect of solid catalyst calcination temperature (673–1033 K) was also studied. Finally, the affection in the weak and moderate acidity for bisphenol F synthesis were also evaluated.
2 Experimental
2.1 Materials
Commercially available montmorillonite K10 as the raw material supplied by Sinopharm Chemical Reagent Co, Ltd, China. Analysis of its mineralogy composition showed it to be 95% montmorillonite. Its anhydrous structural (layer) formula that formed from one octahedral sheet between two tetrahedral sheets 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 layer type. The montmorillonite K10 had a basal (001) spacing of 0.96 nm and the formula of [Si7.86Al0.14][Al2.84Fe0.30Mg0.86]O20(OH)4 according to the literature.14 Montmorillonite K10, which is calcium-rich in commodities form and was converted into the Na-MMT (denoted as MMT) by treatment with NaCl (1 mol L−1 NaCl solution, 100 mL solution per g of clay, 80 °C for 2 h); phenol, formaldehyde (37–40%), aluminum chloride hexahydrate were purchased from Sinopharm Chemical Reagent Co, Ltd, China.
:1 layer type. The montmorillonite K10 had a basal (001) spacing of 0.96 nm and the formula of [Si7.86Al0.14][Al2.84Fe0.30Mg0.86]O20(OH)4 according to the literature.14 Montmorillonite K10, which is calcium-rich in commodities form and was converted into the Na-MMT (denoted as MMT) by treatment with NaCl (1 mol L−1 NaCl solution, 100 mL solution per g of clay, 80 °C for 2 h); phenol, formaldehyde (37–40%), aluminum chloride hexahydrate were purchased from Sinopharm Chemical Reagent Co, Ltd, China.
2.2 Catalyst preparation
A series of Al-MMT(x) were synthesized by aluminum hydroxy oligomeric intercalated in montmorillonite K10 according to previous report15 (x represents the molar of Al in per gram of MMT). Typically, Al-MMT(6) was prepared as follows: (i) preparation of the Al-pillaring agent. 0.1 mol L−1 solution of NaOH was slowly added to 0.1 mol L−1 AlC13 solutions prepared from AlCl3·6H2O to obtain a final hydrolysis ratio OH−/Al3+ = 2.4. Aging at 50 °C for 24 h. (ii) Syntheses of Al-MMT(6). The Al-pillaring agent was slowly added to MMT slurry of 2 g/100 mL (6 mmol of Al per gram of MMT). The final suspension was stirred at room temperature for 24 h, then was transferred to dialysis tubing and dialyzed in distilled water for 3 days to remove C1−. The dialysis water was changed each 24 h until the water-tested C1− free by the AgNO3 test. The resultant mixture was subsequently separated from suspension by filtration to obtain powders, dried in an air and calcined at 400 °C for 4 h. Other samples were prepared as the above procedures.
2.3 Characterization
X-ray diffraction (XRD) was performed on a D8-Advance with a Bruker diffractometer using Cu Kα radiation (40 kV, 40 mA) in the range between 2° and 30° and Cu Kα radiation (40 kV, 40 mA) in the range between 10° and 80°. Diffraction data were recorded with a scanning speed of 0.5° of 2θ s−1 and 2θ = 0.02° at a temperature of 25 °C. The chemical composition of the sample was determined by energy-dispersive X-ray (EDX) analysis using an FEI QUANTA-200. Nitrogen desorption isotherms were performed at 77 K on an ASAP 2010 sorption system (Micromeritics). The samples were degassed at 200 °C for 10 h before measurement. The specific surface area was estimated by the Brunauer–Emmett–Teller (BET) equation. The strength and amount of acid sites in the solid catalysts were estimated by using NH3-TPD measurements with a Micromeritics AutoChem II 2920 V3.05 instrument. Prior to analysis, the catalysts sample were heated from room temperature to 200 °C with a ramp of 10 °C min−1 and purged with helium gas for 30 min. The sample was cooled down to 60 °C under a flow of helium gas and then followed by adsorption of NH3 for 60 min. NH3 is desorbed in helium gas flow from 60 to 700 °C with a ramp of 10 °C min−1 and the signals of NH3-TPD were recorded with an online thermal conducted detector (TCD). Infrared spectra were recorded in the wavelength range of 800–4000 cm−1 using a Bruker vector 22 FT-IR spectrophotometer using KBr disk technique. Pyridine adsorption monitored by in situ infrared FTIR spectra (Py-IR) of the catalyst samples were recorded with a Brucker Vector 22 spectrometer in the absorption mode with a resolution of 4 cm−1. Self-supporting wafers were made and loaded in an IR cell. The wafers were pretreated at 400 °C under flowing oxygen for 2 h, background spectra were recorded after the sample was cooled to room temperature. Adsorption of pyridine was then conducted until saturation. Py-IR spectra were recorded after degassing for 0.5 h.
2.4 Catalytic activity tests
The hydroxyalkylation of phenol with formaldehyde to bisphenol F was performed in a magnetically stirred glass reactor fitted with a reflux condenser and an arrangement for temperature control. In a typical procedure, 82.86 mmol of phenol, 5.73 mmol of formaldehyde and catalyst (0.12 g) were added into the reactor. The reaction mixture was heated to 383 K. After 80 min, the reaction was stopped and cooled down to room temperature, then the solid catalysts were separated by filtration. The products were analyzed by a HPLC system with a Shimadzu LC-20AT system coupled with a SPD-20A UV/Vis detector and a Phenomenex Luna C18 column (250 × 4.6 mm, 5 μm) and column oven temperature was 25 °C, and mobile phase was methanol:water with 65:35 v/v, a flow rate of 0.6 mL min−1. All the reaction conditions followed the above procedures unless otherwise stated.
Performances of MMT, Al-MMT(x), and Al-MMT(6)-Y (Y = 773 K, 923 K, 1033 K) were evaluated in terms of percentage of product yields and percentage of product selectivity, which the equations are defined as follows:
| Yield% = 100 × N/M, selectivity% = 100 × N/F |
where
N is the actual moles of product formed in the reactor,
M denotes expected moles of product formed based on formaldehyde consumed and
F denotes the amount (mol) of all the products.
3 Results and discussion
3.1 Catalyst characterization
The basal spacings of the samples were calculated according to the d001 value measured by XRD (small angle). Fig. 1(A) shows the XRD patterns of MMT and Al-MMT(x) prepared with the different A13+/clay ratio treated at 673 K. The Al-MMT(x) exhibited the 001 reflection at a lower diffraction angle with an increase in A13+/clay ratio, indicating an expansion of the basal spacing of the catalysts. All of the catalysts have a 001 reflections corresponding to basal spacings of approximately 1.7–2.1 nm, which is larger than that of MMT (shows the thickness of the clay layer of 0.96 nm). Note that the A13+/clay ratio is an important factor in the enlargement of the basal spacing of the catalysts. Fig. 1(B) shows wide-angle XRD patterns of MMT and Al-MMT(x). The XRD pattern of all solid catalysts, typical peaks of trioctahedral subgroup of 2:1 phyllosilicates are observed, which are ascribed to (110), (020), (004), (130), (200), (330) and (060) diffractions.16–18 When more Al-pillaring agent were intercalated in the clay layers, all samples did not exhibit any peaks assignable to Al species, suggesting that the Al-pillaring agent was well intercalated into interlayered clay and no long-range change in the crystal structure during the reaction.
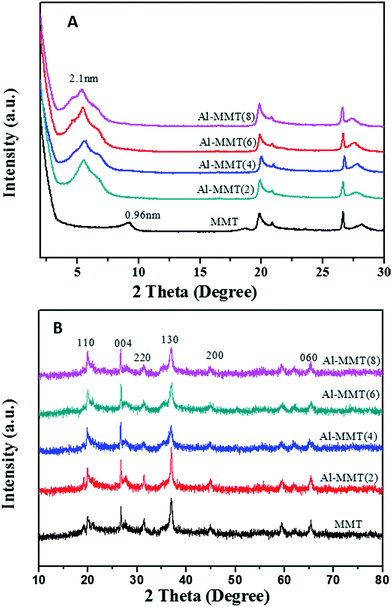 |
| | Fig. 1 XRD patterns of MMT and Al-MMT(x) samples. | |
FT-IR spectra of the catalysts MMT and Al-MMT(x) are presented in Fig. 2. It can be clearly seen from Fig. 2 that the bands appearance around 3448 cm−1 and 1640 cm−1 are assigned to the stretching and bending vibration of the OH group in water molecules.19 In all curve, the band located at 1033 cm−1 was indexed to the asymmetric stretching bonds of Si–O–Si, associated with the motion of oxygen in Si–O–Si antisymmetric stretching.15,20 The band at around 3640 cm−1 is due to the vibration of Al–OH, and the absorption at 849 cm−1 is due to the Si–OH in the tetrahedral layer.21 Besides, significant decreases of the peaks intensity near 3448 cm−1 and 1640 cm−1 with increase Al-pillaring agent, which means a decrease of water content in the interlayer space.22 Compared to the FT-IR spectra of pure MMT, significant increase of the peaks intensity at 3642 cm−1 with increasing content of Al-pillaring agent, which is attributable to the appearance of Al–OH bending vibration and the intensity is also enhanced. The above results revealed that Al-pillaring agent were successfully intercalated into the interlayer of MMT.
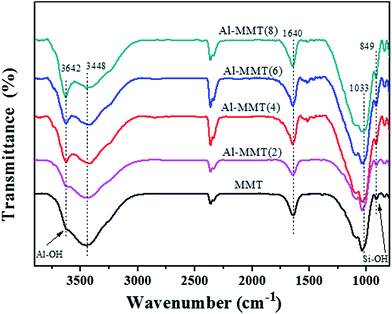 |
| | Fig. 2 FT-IR spectra of MMT and Al-MMT(x) samples. | |
The profiles of NH3-TPD to characterize acidic properties of MMT and Al-MMT(x) are shown in Fig. 3. The amount of surface acid sites based on NH3 desorption are presented in Table 2. As shown in Fig. 3, the solid catalyst showed three types of desorption peaks from 90 °C to 700 °C region which could be assigned to weak, moderate and strong acid sites.23–25 The weak and moderate acid sites on MMT or Al-MMT(x) is related to the amount of montmorillonite interlayer aluminum ions, which engender sites of weak and moderate acidity in the form of the OH groups bonded to the pillars Al ions (Al–OH). Meanwhile, the strong acid sites is associated with the OH groups bonded to tetrahedrally coordinated Al ions (AlTd–OH).26–28 Interestingly, the total amount of acid sites of Al-MMT(x) increase obviously with increasing Al/clay ratio from 2 to 6. At the same time, the amount of weak and moderate acid sites of Al-MMT(x) enhanced considerably from 0.29 to 0.38 mmol g−1 and from 0.58 to 1.41 mmol g−1, respectively. However, the amount of strong acid sites decreased from 2.14 to 1.88 mmol g−1. When further increase the A13+/clay ratio up to 8, increased slightly in the amount of surface acid sites was observed. This is because the decrease in the specific surface areas, which is closely related to the amount of surface acid sites. The acidic property of the catalyst is a key factor for hydroxyalkylation of phenol with formaldehyde to bisphenol F.
 |
| | Fig. 3 NH3-TPD profiles of MMT and Al-MMT(x) samples. | |
The acidic properties were also studied by Py-IR spectroscopy to evaluate the Lewis and Brönsted character. The spectra are presented in Fig. 4 and the amount of the Lewis and Brönsted acid sites are shown in Table 2. Two types of adsorbed pyridine species were observed. The band at 1540 cm−1 can be assigned to Brönsted acid sites and the band at 14480 cm−1 can be attributed to Lewis acid sites according to the literatures.29,30 While the band at 1490 cm−1 originated from the coadsorption of pyridine on both Brönsted and Lewis acid sites. The Al-pillaring agent significantly affected the acidity of the MMT materials. Fig. 4 clearly shows that both the concentration of Brönsted and Lewis acid sites increase, and the L/B ratio increases almost linearly with increasing Al-pillaring agent, which indicates that the addition of Al-pillaring agent resulted in the redistribution of the two types of acid sites. This result is well consistent with the above NH3-TPD result.
 |
| | Fig. 4 Py-IR spectra of MMT and Al-MMT(x) samples. | |
The specific surface areas of MMT and Al-MMT(x) samples are presented in Table 1. Among various solid catalysts, MMT showed the lowest (60 m2 g−1) specific surface areas. The specific surface areas of Al-MMT(x) shows a significantly increase compared with parent MMT, which is because the expansion of the basal spacing of clay layers after being intercalated by Al-pillaring agent. The solid catalysts were found to be in the sequence of Al-MMT(6) > Al-MMT(8) > Al-MMT(4) > Al-MMT(2) > MMT. Compared with other samples, Al-MMT(8) showed decrease in the specific surface areas. This is due to the excess Al-pillaring agent occupied the spaces of gallery channels and blocked the pore of the clay layers.
Table 1 Textural properties and elemental composition of various solid acid catalysts
| Catalyst |
BET surface area (m2 g−1) |
Composition |
| Si (wt%) |
O (wt%) |
Mg (wt%) |
Al (wt%) |
| MMT |
61 |
36.5 |
57.9 |
0.7 |
15.8 |
| Al-MMT(2) |
115 |
34.9 |
56.7 |
0.5 |
18.7 |
| Al-MMT(4) |
169 |
34.6 |
53.3 |
0.6 |
21.4 |
| Al-MMT(6) |
207 |
33.7 |
51.1 |
0.5 |
24.9 |
| Al-MMT(8) |
194 |
34.4 |
50.8 |
0.6 |
27.3 |
3.2 Activity measurement
Activity of MMT and Al-MMT(x) screening experiments for the hydroxyalkylation of phenol were be implemented and the results are presented in Table 3. The yield of bisphenol F significantly rose with the presence of aluminum species, confirming that catalytic activity is a result of aluminum species presence in the MMT. Among various solid catalysts, the MMT showed the least activity (10% yield), there could be due to its the lowest specific surface area and presence of the lowest acidity (Table 2). As compared to MMT, Al-MMT(x) showed higher product yield due to its higher acidity.31 The bisphenol F yield increasing from 10.6 to 89.2% with increase in A13+/clay ratio from 2 to 6. This is attribute to enhanced amount of weak and moderate acid sites and higher L/B ratio resulted in higher catalytic activity, which are confirmed by the results of NH3-TPD and Py-IR (Tables 2 and 3). When further increase A13+/clay ratio up to 8, there is a decrease of bisphenol F yield and selectivity (yield% = 87.3, sel.% = 90.4). Generally, a higher Al-pillaring agent content means a higher amount of both weak and moderate acid sites as well as higher specific surface areas. However, a further increase of A13+/clay ratio gives rise to a decline of specific surface areas, and slight increase in weak and moderate acid sites. In other words, excess Al-pillaring agent occupied the spaces of gallery channels and blocked the pore of clay layers, and results in decline of specific surface areas. As can be seen from NH3-TPD characterization of various catalysts and the results of catalytic reaction showed that both weak and moderate acid sites of Al-MMT(x) are critical for achieving the highest yield of bisphenol F in hydroxyalkylation of phenol.
Table 2 Acidic properties of various solid acid catalysts
| Acidity by type |
| Catalyst |
Brönsted (mmol g−1) |
Lewis (mmol g−1) |
Lewis/Brönsted (L/B) |
| MMT |
0.16 |
0.14 |
0.87 |
| Al-MMT(2) |
0.24 |
0.22 |
0.92 |
| Al-MMT(4) |
0.26 |
0.25 |
0.10 |
| Al-MMT(6) |
0.27 |
0.32 |
1.18 |
| Al-MMT(8) |
0.39 |
0.48 |
1.23 |
Table 3 Catalytic activities of Al-pillared clay for bisphenol Fa
| Catalyst |
NH3-TPD amount of acidic sites (mmol g−1) |
Yield/% |
Selectivity/% |
Isomer distribution/% |
| Total |
Weak |
Moderate |
Strong |
4,4′-Isomer |
2,4′-Isomer |
2,2′-Isomer |
| Reaction conditions: phenol/formaldehyde molar ratio, 15:1; catalyst concentration, 0.030 g g−1; reaction temperature, 383 K; reaction time, 80 min. |
| MMT |
2.67 |
0.22 |
0.37 |
2.08 |
10.8 |
90.3 |
17.2 |
52.5 |
30.3 |
| Al-MMT(2) |
3.02 |
0.29 |
0.58 |
2.14 |
69.2 |
92.2 |
20.5 |
51.1 |
28.4 |
| Al-MMT(4) |
3.27 |
0.35 |
0.97 |
1.94 |
74.5 |
91.3 |
31.2 |
49.2 |
19.6 |
| Al-MMT(6) |
3.70 |
0.41 |
1.41 |
1.88 |
89.2 |
92.7 |
30.5 |
50.4 |
19.1 |
| Al-MMT(8) |
3.74 |
0.46 |
1.52 |
1.76 |
87.3 |
90.4 |
32.2 |
48.5 |
19.3 |
| Al-MMT(6)-773 |
3.27 |
0.30 |
1.08 |
1.89 |
80.2 |
91.6 |
30.1 |
49.3 |
20.6 |
| Al-MMT(6)-923 |
3.21 |
0.25 |
0.86 |
2.10 |
70.4 |
92.2 |
26.4 |
44.2 |
29.4 |
| Al-MMT(6)-1033 |
3.01 |
0.21 |
0.53 |
2.27 |
66.8 |
91.5 |
25.6 |
46.4 |
28.0 |
3.3 Effect of catalyst calcination temperature
Calcination temperature is closely related to the interlayer space height and the amount of surface acid sites. Therefore, the effect of catalyst calcination temperature was investigated. The Al-MMT(6) catalysts calcined at 673, 773, 923, and 1033 K were used in the hydroxyalkylation of phenol to bisphenol F, and the results are shown in Table 3. The yield of bisphenol F decreased from 89.2 to 66.8% with calcination temperature is increased from 673 to 1033 K. The reason for this result may be that the decrease of the surface acid sites and collapse of clay layers. To confirm the above speculation, the catalysts calcined at 673, 773, 923, and 1033 K were subsequently explored using NH3-TPD, XRD, and FT-IR. NH3-TPD patterns of catalyst calcined at 673, 773, 923, and 1033 K are shown in Fig. 5(A). After calcination temperature increased from 673 to 1033 K, the weak and moderate acid sites peak intensity were diminished. This is due to Al–OH⋯O–Si–hydrogen bonds were replaced during thermal treatments by covalent bonds that form Al–O–Si linkages between the pillars and the clay layers.32 Hence, it lead to the decrease of surface acid sites.
 |
| | Fig. 5 (A) NH3-TPD profile and (B) FT-IR spectra of Al-MMT(6) at different calcination temperatures (673 K, 773 K, 923 K, 1033 K). | |
Furthermore, FT-IR spectra of these catalysts are presented in Fig. 5(B). It could be clearly found that significant decrease of the peak intensity at 3642 cm−1 (corresponding to the Al–OH) with calcination temperature increased from 673 to 1033 K, which is attributed to decomposition of the Al-polyoxocations at higher temperatures.33 Fig. 6 shows the XRD patterns of the Al-MMT(6) calcined at 673, 773, 923, and 1033 K. For catalysts calcined at 773 K and 923 K, exhibits widening and decreasing intensity, 001 reflection peak. Moreover, the 001 reflection peak is shifted toward higher diffraction angles after calcination at 1033 K. These results indicate that the crystalline structure of the clay layer has been destroyed and clay layers appeared collapse.34
 |
| | Fig. 6 XRD patterns of Al-MMT(6) at different calcination temperatures (673 K, 773 K, 923 K, 1033 K). | |
3.4 Optimization of reaction parameters
The effects of phenol/formaldehyde ratio on product yield and bisphenol F selectivity for Al-MMT(6) catalyst are illustrated in Fig. 7; the product yield is increased significantly from 33 to 85% with increase in the phenol/formaldehyde ratio from 5 to 15; further increase in mole ratio up to 20 does not result in any significant changes. The lower product yield was mainly because of the further reaction of bisphenol F with formaldehyde to give tarry products such as trimers and higher homologues under the condition of low phenol/formaldehyde ratio.35 The 4,4′-isomer increased from 27 to 30% and 2,2′-isomers decreased from 25 to 21%, whereas the amounts of 2,4′- isomers remained almost unchanged, with increase in phenol/formaldehyde ratio from 5 to 20. Thus, the optimal molar ratio was 15.
 |
| | Fig. 7 Effect of phenol/formaldehyde molar ratio on yield, selectivity and isomers' distribution of bisphenol F. Reaction conditions: catalyst, Al-MMT(6); reaction time, 80 min; reaction temperature, 383 K; catalyst concentration, 0.030 g g−1. | |
The effects of reaction temperature on product yield, bisphenol F selectivity and isomer distribution over Al-MMT(6) was investigated and shown in Fig. 8. It was clearly observed that the product yield significantly increased from 36 to 89.2%, while remained almost unchanged in bisphenol F selectivity with increased temperature from 343 to 383 K. The amount of 4,4′-isomer decreased from 49 to 32%, however, the amount of 2,4′- and 2,2′-isomers increased from 42 to 47% and 9 to 21%, respectively. It was demonstrated that high temperatures is needed for hydroxyalkylation of phenol with formaldehyde to promote the formation of bisphenol F. However, if further increase in temperature, formaldehyde to evaporate from the mixture, which is not conducive reaction proceeds. Thus, 383 K was chosen as the optimal reaction temperature.
 |
| | Fig. 8 Effect of reaction temperature on the yield, selectivity and isomers' distribution of bisphenol F. Reaction conditions: catalyst, Al-MMT(6); mole ratio of phenol/formaldehyde, 15; reaction time, 80 min; reaction temperature, 343–383 K; catalyst concentration, 0.030 g g−1. | |
Results showing the effects of the catalyst concentration on product yield, selectivity to bisphenol F and isomer distribution are presented in Fig. 9. The product yield increased markedly from 77 to 88% as the catalyst concentration increased from 0.01 to 0.030 g g−1. When further increase in catalyst concentration, the product yield and selectivity to bisphenol F only shows a minor change. This is expected due to the bisphenol F conversion into a trimer as higher catalyst concentration. The amount of 4,4′-isomer increased from 25 to 28%, while no significant change was observed in the amount of 2,2′- and 2,4′-isomers, as increase the catalyst concentration from 0.01 to 0.04 g g−1. Thus, 0.030 g g−1 was selected as the suitable catalyst dose.
 |
| | Fig. 9 Effect of catalyst concentration on the yield, selectivity and isomers' distribution of bisphenol F. Reaction conditions: catalyst, Al-MMT(6); mole ratio of phenol/formaldehyde, 15; catalyst concentration, 0.010–0.040 g g−1; reaction time, 80 min; reaction temperature, 383 K. | |
Fig. 10 depicts the effects of reaction time on product yield, bisphenol F selectivity and its isomer distribution. The results shows that the product yield increased from 46 to 88% with increase in reaction time from 20 to 90 min. No significant change was discovered in the selectivity of bisphenol F over the entire range of the reaction course; there is a slight change for the distributions of isomer during the whole reaction progress. Based on the results, we consider that 80 min is the reaction time for optimal performance.
 |
| | Fig. 10 Effect of reaction time on the yield, selectivity and isomers' distribution of bisphenol F. Reaction conditions: catalyst, Al-MMT(6); mole ratio of phenol/formaldehyde, 15; reaction time, 20–90 min; catalyst concentration, 0.030 g g−1. | |
The catalyst stability of Al-MMT(6) was tested by performing recycle experiments, which were carried out at 383 K with phenol to formaldehyde mole ratio of 15. The testing results are presented in Fig. 11, which the activity of the catalyst shows no significant change even after four successive runs showing 84.3% product yield and 91.5% bisphenol F selectivity. The slight decrease of the product yield could be because of the leaching out of the Al-pillaring agent from the clay interlayers and losses of the catalyst during reuse. Although, the synthesized Al-MMT(6) shown excellent stability, which was greatly important to its practical applications.
 |
| | Fig. 11 Reusability of catalyst study. Reaction conditions: catalyst, Al-MMT(6); mole ratio of phenol/formaldehyde, 15; catalyst concentration, 0.030 g g−1; reaction temperature, 383 K; reaction time, 80 min. | |
3.5 Plausible mechanistic pathway
A proposed mechanistic pathway to illustrate the processes of phenol with formaldehyde to give bisphenol F by Al-MMT(6) is shown in Scheme 1. Indeed, it has been shown that such contact with an acid catalyst.36 The monomeric form of formaldehyde that presents in aqueous solution is mostly in the form of methylene glycol and oligomer. In the first step, the protonation of formaldehyde is activated by Brönsted acid sites of the catalyst to form the CH2OH+, then converted to carbocations. At the same time, the carbocations were produced by the abstraction of H− over Lewis acid sites of the catalyst. Then, the molecules of hydroxy benzyl alcohol were synthesized with the carbocations may attack phenol. Next, the hydroxymethyl group of the o/p-hydroxy benzyl alcohol abstracts a proton from the catalysts to form hydroxy benzyl carbocation. Ultimately, the bisphenol F may be formed as the electrophilic attack of hydroxy benzyl carbocation on the second molecule of phenol followed by a proton transfer. The hydroxyalkylation of phenol to bisphenol F is catalyzed by the synergy of Brönsted acid sties and Lewis acid sties. This result was confirmed by Py-IR.
 |
| | Scheme 1 Proposed plausible mechanism for hydroxyalkylation of phenol and formaldehyde to bisphenol F. | |
4 Conclusions
We have prepared a series of Al-MMT(x) composites with A13+/clay ratio from 2 to 8 and tested them for the hydroxyalkylation of phenol. Among the various solid acid catalysts, Al-MMT(6) exhibited the highest catalytic activity for the hydroxyalkylation of phenol, because its weak and moderate acid sites and larger specific surface areas. Moreover, the effect of catalyst calcination on the amount of weak and moderate acid sites was studied, the results showed that both weak and moderate acid sites quantity appears the reduction under the high temperature. The catalytic performance of these catalysts depended on weak and moderate acidity and the textural properties (specific surface areas). In addition, the effects of various reaction parameters like mole ratio, catalyst concentration, reaction temperature and time on product yield and the selectivity to bisphenol F were investigated, and an 89.2% yield and 92.7% selectivity to bisphenol F was obtained under optimal conditions. Finally, the catalyst was found to retain its activity even after a fourth recycle experiment.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (no. 51378187, J1210040), Innovative Research Team Development Plan-Ministry of Education of China (no. IRT1238), the Key Project of Hunan Provincial Education Department (no. 13CY001), and Hunan Provincial International Cooperation Project of China (no. 2014WK3030).
Notes and references
- Y. Jiao, L. Ding, S. Fu, S. Zhu, H. Li and L. Wang, Anal. Methods, 2012, 4, 291–298 RSC.
- E. Danzl, K. Sei, S. Soda, M. Ike and M. Fujita, Int. J. Environ. Res. Public Health, 2009, 6, 1472–1484 CrossRef CAS PubMed.
- X. Zhang, Q. He, H. Gu, H. A. Colorado, S. Wei and Z. Guo, ACS Appl. Mater. Interfaces, 2013, 5, 898–910 CAS.
- M. Okihama and J. Kunitake, Patent JP 08198790, 1996.
- M. Okihama and J. Kunitake, Patent JP 08268943, 1996.
- M. Okihama and J. Kunitake, Patent JP 09067287, 1997.
- W. Wang, Y. Yang, J. Bao and Z. Chen, J. Fuel Chem. Technol., 2009, 37, 701–706 CrossRef.
- K. Nowinska and W. Kaleta, Appl. Catal., A, 2000, 203, 91–100 CrossRef CAS.
- A. Pacuła, K. Pamin, J. Kryściak-Czerwenka, Z. Olejniczak, B. Gil, E. Bielańska, R. Dula, E. M. Serwicka and A. Drelinkiewicz, Appl. Catal., A, 2015, 498, 192–204 CrossRef.
- M. Chen, J. Yan, Y. Tan, Y. Li, Z. Wu, L. Pan and Y. Liu, Ind. Eng. Chem. Res., 2015, 54, 11804–11813 CrossRef CAS.
- S. K. Jana, T. Kugita and S. Namba, Catal. Lett., 2003, 90, 3–4 CrossRef.
- A. Jha, A. C. Garade, S. P. Mirajkar and C. V. Rode, Ind. Eng. Chem. Res., 2012, 51, 3916–3922 CrossRef CAS.
- A. C. Garade, V. S. Kshirsagar, R. B. Mane, A. A. Ghalwadkar, U. D. Joshi and C. V. Rode, Appl. Clay Sci., 2010, 48, 164–170 CrossRef CAS.
- S. Yang, G. Liang, A. Gu and H. Mao, Ind. Eng. Chem. Res., 2012, 51, 15593–15600 CrossRef CAS.
- A. Gil, R. Trujillano, M. A. Vicente and S. A. Korili, Adsorpt. Sci. Technol., 2007, 25, 3–4 CrossRef.
- A. Pacuła, K. Pamin a, J. K. Czerwenka, Z. Olejniczak, B. Gil, E. Bielanska, R. Dula, E. M. Serwicka and A. Drelinkiewicz, Appl. Catal., A, 2015, 498, 192–204 CrossRef.
- S. Yang, G. Liang, A. Gu and H. Mao, Mater. Res. Bull., 2013, 48, 3948–3954 CrossRef CAS.
- H. Mao, B. Li, X. Li, Z. Liu and W. Ma, Catal. Commun., 2009, 10, 975–980 CrossRef CAS.
- M. Sulpizi, M. P. Gaigeot and M. Sprik, J. Chem. Theory Comput., 2012, 8, 1037–1047 CrossRef CAS PubMed.
- S.-H. Wu, J.-L. Wu, S.-Y. Jia, Q.-W. Chang, H.-T. Ren and Y. Liu, Appl. Surf. Sci., 2013, 287, 389–396 CrossRef CAS.
- H. Mao, B. Li, X. Li, Z. Liu and W. Ma, Mater. Res. Bull., 2009, 44, 1569–1575 CrossRef CAS.
- M. S. Marth, K. L. Walther and A. Wokaun, J. Non-Cryst. Solids, 1992, 143, 93–111 CrossRef.
- S. Shylesh, A. Wagener, A. Seifert, S. Ernst and W. R. Thiel, ChemCatChem, 2010, 2, 1231–1234 CrossRef CAS.
- K. C. Park and S. K. Ihm, Appl. Catal., A, 2000, 203, 201–209 CrossRef CAS.
- M. Höchtl, A. Jentys and H. Vinek, Appl. Catal., A, 2001, 207, 397–405 CrossRef.
- S. S. M. Konda, S. Caratzoulas and D. G. Vlachos, ACS Catal., 2016, 6, 123–133 CrossRef CAS.
- H. Knözinger and P. Ratnasamy, Catal. Rev.: Sci. Eng., 1978, 17, 31–70 Search PubMed.
- M. N. Timofeeva, V. N. Panchenko, M. M. Matrosova, A. S. Andreev, S. V. Tsybulya, A. Gil and M. A. Vicente, Ind. Eng. Chem. Res., 2014, 53, 13565–13574 CrossRef CAS.
- F. Chambon, F. Rataboul, C. Pinel, A. Cabiac, E. Guillon and N. Essayem, Appl. Catal., B, 2011, 105, 171–181 CrossRef CAS.
- H. Zhou, W. Zhu, L. Shi, H. Liu, S. Liu, S. Xu, Y. Ni, Y. Liu, L. Li and Z. Liu, Catal. Sci. Technol., 2015, 5, 1961–1968 CAS.
- I. A. Rahman, P. Vejayakumaran, C. S. Sipaut, J. Ismail and M. A. Bakar, Colloids Surf., A, 2007, 294, 102–110 CrossRef CAS.
- M. L. Occelli, J. A. Bertrand, S. A. C. Gould and J. M. Dominguez, Microporous Mesoporous Mater., 2000, 34, 195–206 CrossRef CAS.
- A. Sampieri, G. Fetter, P. Bosch and S. Bulbulian, Langmuir, 2006, 22, 385–388 CrossRef CAS PubMed.
- Z. Ge, D. Li and T. J. Pinnavaia, Microporous Mater., 1994, 3, 165–175 CrossRef CAS.
- R. Liu, X. Xia, X. Niu, G. Zhang, Y. Lu, R. Jiang and S. He, Appl. Clay Sci., 2015, 105–106, 71–77 CrossRef CAS.
- R. Liu, X. Niu, X. Xia, Z. Zeng, G. Zhang and Y. Lu, RSC Adv., 2015, 5, 62394–62401 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 











