
The synthesis and comparison of chondroitin sulfate-modified PDMAEMA with chondroitin sulfate-modified PEI as a potential gene delivery vector†
Abstract
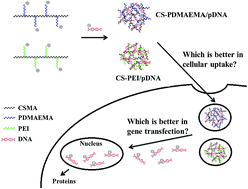
Our previous research has confirmed a chondroitin sulfate-polyethylenimine copolymer (CS-PEI) as a potential gene delivery vector because of its recognition by CD44, which enhances the cellular uptake of CS-PEI/pDNA polyplexes. As poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) is also a commonly used non-viral gene delivery vector, a CS-PDMAEMA copolymer was synthesized using a similar method with CS-PEI via Michael addition. The physicochemical properties of CS-PDMAEMA and CS-PEI copolymers were thoroughly characterized. The gel electrophoresis results demonstrate that the weight ratio of CS-PEI/pDNA required to completely encapsulate pDNA is ≥1 while that of CS-PDMAEMA/pDNA is ≥3. The CS-modified cationic polymers show lower cytotoxicity than compared with the unmodified ones. At the same weight ratio, CS-PEI/pDNA has a smaller particle size than CS-PDMAEMA/pDNA. The cellular uptake of CS-modified polyplexes is higher in U87 cells (high CD44 expression) than in 3T3 cells (low CD44 expression). However, the transfection efficiency of CS-modified polyplexes is higher in 3T3 cells than in U87 cells. The contrasting results may be attributed to the variation of cell types. In addition, the high level of asialoglycoprotein receptor (ASGP-R) expressed in 3T3 cells seems beneficial for triggering the lectin receptor-mediated endocytosis and results in high transfection efficiency when compared with U87 cells.
 Please wait while we load your content...
Please wait while we load your content...

