
Template-free synthesis of 3D hierarchical nanostructured NiCo2O4 mesoporous ultrathin nanosheet hollow microspheres for excellent methanol electrooxidation and supercapacitors†
Abstract
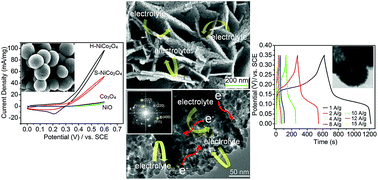
A facile template-free solvothermal method without any additional alkali was developed to fabricate a 3D hierarchical hollow microsphere precursor, followed by annealing in air, leading to a novel 3D hierarchical NiCo2O4 hollow microsphere material, which is composed of mesoporous (16.1 nm) ultrathin nanosheets (∼11–21 nm) consisting of ultrafine NiCo2O4 nanoparticles (11.9 nm). This 3D hierarchical NiCo2O4 nanosheet hollow microsphere material possesses a high specific surface area (93.4 m2 g−1) and mesoporosity, and thus superior electrochemical performance as an advanced electrode material. For methanol electrooxidation, the 3D hierarchical NiCo2O4 nanosheet hollow microsphere displays much higher electrocatalytic activity (95 A g−1, at 0.6 V), lower overpotential (0.27 V, vs. SCE), and higher stability compared with the 3D hierarchical NiCo2O4 nanosheet solid microspheres, Co3O4 and NiO microspheres. For supercapacitors, the NiCo2O4 hollow microsphere exhibits excellent specific capacitance of 1701 F g−1 at 1 A g−1, excellent rate capability (61.5% retention at 15 A g−1), and good electrochemical stability with 78.2% retention after 1000 charge–discharge cycles even at a high current density of 10 A g−1. These findings can be explained by the unique integral characteristics of 3D NiCo2O4 hollow spheres with high electron conductivity, large surface area and numerous open spaces between neighboring mesoporous ultrathin nanosheets, which can offer many facile diffusion paths for ion/electrolyte and greatly improve the electron/ion transfer within the electrode and at the electrode–electrolyte interfaces.
 Please wait while we load your content...
Please wait while we load your content...

