
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimerization and ligand binding in tyrosylprotein sulfotransferase-2 are influenced by molecular motions†
Warispreet Singha,
Tatyana G. Karabencheva-Christova*a,
Olivier Sparaganob,
Gary W. Blacka,
Petar Y. Petrovc and
Christo Z. Christova
aDepartment of Applied Sciences, Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK. E-mail: christo.christov@northumbria.ac.uk; tatyana.karabencheva-christova@northumbria.ac.uk; Tel: +44 (0)191 243 7964, +44 (0)191 243 4077
bVice Chancellor's Office, Coventry University, Coventry, CV1 5FB, UK
cDepartment of Organic Chemistry, Faculty of Chemistry and Pharmacy, Sofia University “St Kliment Ohridski”, Sofia, Bulgaria
First published on 2nd February 2016
Abstract
Tyrosylprotein sulfotransferase-2 catalyses important, but a less explored posttranslational modification of proteins. The crystallographic structure of the full complex, containing the enzyme, PAPS cofactor and the peptide substrate (WTFC) revealed important structural and atomistic details about the overall structure, binding sites and interactions in TPST-2, but is not informative about the conformational flexibility, which is a fundamental protein property. In a recent computational study we analyzed the impact of conformational flexibility on key geometric determinants and interactions in WTFC. However, there are still unexplained effects about how the conformational dynamics influences the formation of the catalytically active dimer form of the enzyme and also how the binding of the cofactor and the substrate to the apoenzyme influences the structure and the pattern of correlated motions in the enzyme. In order to provide the missing knowledge we performed a molecular dynamics study on the dimerization mutant W113A, the apoenzyme (APO), enzyme–cofactor complex (ES), and enzyme–substrate complex (ES) and compared the results to the WTFC. We identified new hydrophobic interactions important for the stabilization of the enzyme dimer and provided structural insight about the role of the key mutation W113A for the dimerization in the absence of experimentally-derived structure. In addition we showed that the binding of the substrate and cofactor to the apoenzyme contributes to the stability of the whole active complex, influences the local interactions in the binding site and importantly, affects the pattern of the correlated motions in the entire molecule.
Introduction
Sulfation of tyrosine residues in proteins is a very important post synthetic modification, which is still explored little. The reaction is catalyzed by an enzyme – Tyrosylprotein sulfotransferase (TPST).1,2 Tyrosine sulfation is important for cell signaling and is also target for drug design.3,4 Sulfated tyrosine residues are important for the interaction of HIV's gp120 protein with CCR5 in order to get entry to CD4+ T-lymphocytes.5 Sulfated PSGL-1 is involved in the infection process of enterovirus 71 virus, which is responsible for neurological diseases in children.6 Sulfated tyrosine residues are found in approximately sixty immune system proteins.7,8 The sulfation of protein tyrosine residues is carried out as enzyme catalyzed transfer of negatively charged sulfate group from the sulfate donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to the hydroxyl group of a tyrosine leading to a tyrosine O4-sulfate ester and adenosine 3′,5'-diphosphate.2 Two isoforms TPTST-1 and TPST-2, exist in humans.9 TPST has a type II transmembrane topology i.e. a short N-terminal cytoplasmic domain, a 17 residue transmembrane domain, and a luminal catalytic domain.10 The enzyme has two N-glycosylation sites, that are four cysteine residues on the luminal oriented side of enzyme.4 The crystal structure of the catalytic domain of human TPST-2 (denoted as TPST2ΔC18, containing the sequence from G43 to L359) in complex with PAP (3′-phosphoadenosine 5′-phosphate) and the substrate (the peptide C4P5Y3) was solved at a resolution of 1.9 Å11 (Fig. 1). The substrate peptide contains only one tyrosine residue which is a sulfate acceptor and is surrounded by six acidic residues, which provide the substrate negative charge of minus six.11 The catalytic domain of TPTS-2, consists of a α/β motif with a five-stranded parallel β-sheet, surrounded by α helices.11 The TPST-2 exists as a homodimer, which is the catalytically active state of the enzyme, where two subunits of the dimer are designated as protomer A and protomer B11 and both participate in the formation of the binding site and the catalytically active complex. The alpha helices α2–α4 play an important role in dimer formation in TPST-2 and residues R118 and R122 harboring α2–α3 also participate in the substrate binding of the other protomer and vice versa (Fig. 1). Residue W113 plays a key role in the dimerization process and W113A mutant appears to exist as monomer in solution based on experimental studies.12 The crystal structure of human TPST-2,12 provides important knowledge about the overall fold, atomistic interactions and contacts between its both protomers, however it shows no information about how conformational flexibility affects key interactions responsible for the formation of the catalytically active dimer and the structural changes, associated with the binding of the ligands (cofactor and substrate). Molecular dynamics is one of the most appropriate and broadly implemented methods for studying protein flexibility and dynamics which provides atomistic insights that cannot be obtained experimentally.13–18 In a recently accepted paper16 we comprehensively analyzed the conformational flexibility of the wild type full complex of TPST2 (WTFC), which includes the enzyme, substrate and the cofactor in their catalytically active dimer form. We studied the mutations of key residues in the substrate binding site and the catalytic site and analyzed the correlated motions in the WTFC.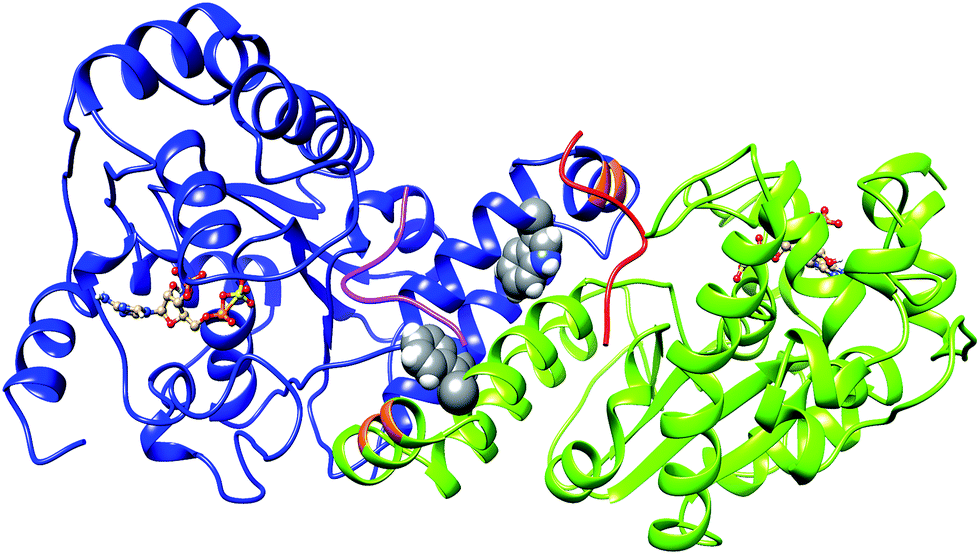 | ||
| Fig. 1 The 3D structure of human TPTS-2 (PDB code: 3AP1) enzyme.12 The cofactor PAPS is represented in ball and stick representation. The protomer A and B of homodimer TPST-2 are shown in deep-cued, round ribbon representation in blue and lime green colors respectively. The substrate peptide of protomer A and B are shown in pink and red colors respectively. The residue R118 and R112 of protomer A participating in substrate binding of protomer B are shown in orange color. The W113 residue involved in dimerization is shown in sphere representation on both protomers. | ||
However, it is essential to understand the atomistic nature of the structural changes which occur upon the binding of each ligand, PAPS and C4P5Y3. MD simulations can provide the needed structural insights, as a fast alternative to the experimentally determined structures, as far as the only available crystal structure so far is the one of the full complex (WTFC).12 In addition it is very important to understand atomistic details of the conformational effects associated with the formation of the protein dimer, which is the catalytically active form of TPST-2. Computational analysis of the effect of W113A mutation can provide unique atomistic insights into the dimerization process. As far as there is no available experimentally derived structure of this key mutant, atomistic MD can be reliable and fast alternative to gain the vital missing structural information. In order to complete the missing knowledge, we extended our study16 and made simulations to answer the following questions: (i) how the conformational flexibility influences the protein–protein interactions in the dimer formation; (ii) what is the atomistic nature of W113A which is a key mutation preventing the dimerization (iii) how does cofactor and substrate binding influence the structure of the apoenzyme (APO), the enzyme with bound cofactor complex (EC) and the enzyme with bound substrate complex (ES); (iv) how does the correlated motions in TPST2 change in the different states of the enzyme upon binding of the ligands.
Methods and computational details
The conformational flexibility of different states of TPST-2 enzyme (APO, EC, ES and W113A mutant) was explored using atomistic molecular dynamics (MD) simulations for 100 ns, by means of Gromacs 4.5.5 package19–21 with GROMOS96 43a1 (ref. 22) force field (Table S1†). The system setup and preparation was done as in ref. 16. The crystal structure of TPST-2 (ref. 12) with PDB code 3AP1 was used. Energy minimization in gas phase was performed by using the steepest descent23 and conjugate gradient24 algorithm until the maximum force was smaller than 100 kJ mol−1 nm−1. Subsequently a periodic box was defined and periodic boundary conditions were applied. The box size was set to 1.0 nm by using Single Point Charge (SPC),25 water model was used to solvate the system and Na+ and Cl− ions were used to neutralize its total charge (Table S1†). New energy minimization was performed for the whole system using again the steepest descent and the conjugate gradient algorithms. After that position restrain molecular dynamics was carried out for 50 ps in NVT ensemble26 at constant temperature of 300 K with time step of 0.002 ps. The productive 100 ns MD simulations were performed in NPT ensemble at constant temperature of 300 K with initial velocities taken from Maxwell velocity distribution at 300 K and an integration time step of 0.002 ps. The system was kept at 300 K. Berendsen temperature coupling and Parrinello–Rahman pressure coupling were with constant (τT) of 0.1 ps and 1 bar pressure, time constant (τP) of 0.5 ps. Electrostatic interactions were treated by the Particle Mesh Ewald (PME).27 The cut off for the Coulomb interactions was 1.0 nm, the Fourier spacing was 0.135 nm. The van der Waals interactions were modelled using Lennard-Jones potential with cut off distance set to 1.4 nm (rvdw) and using switching function. The LINCS algorithm28 was used to maintain all the covalent bonds involving hydrogen atoms rigid. The coordinates were saved every 20 ps.Analysis of molecular dynamics simulations
The analysis of the trajectories obtained from the simulations such as root mean square deviation (RMSD) of Cα atoms of the protein with respect to minimized crystal structure, root mean square fluctuations (RMSF), radius of gyration (Rg), electrostatic interactions, hydrogen bonds, solvent accessible surface area (SASA), and cluster analysis were carried out with the Gromacs tools. The visualization of molecular structures and trajectories was done using VMD29 software. The domain cross-correlation analysis (DCCA) was performed using the Bio3D package.30 The Cij matrix were used to represent a cross correlation between the ith and jth atoms and ranges from −1 to +1. To create the cross correlation Cij matrix the Cα atoms of the protein, substrate and all atoms of the cofactor were included. The positive values in DCCA map reflect the correlated motions and the negative values reflect the anticorrelated motions.Results and discussion
Interactions between the protomers and dimer formation
The equilibration of the MD simulations done for the present analysis has been discussed extensively.16 The TPST-2 enzyme exists as a dimer both in the crystal structure and in vivo.9 The formation of the substrate binding site in WTFC is a result of the dimerization with the participation of α2, α3 and α4 helices. During the simulations, the aliphatic parts of the side chains of R110 and R105 make hydrophobic interactions with the aromatic ring of W113 (Fig. S1†) with average distance of 3.8 Å and 4.1 Å respectively for 100 ns in WTFC. These interactions are a result from conformational flexibility and are not seen in the crystal structure. The side chains of M109, L133 and I102 also make hydrophobic interactions (Fig. S31†) with average distance of 3.8 Å, 4.0 Å and 4.4 Å respectively with the aromatic ring of W113 (Fig. 2). In order to understand the structural effect of a key mutation for the dimerization – W113A, we performed 100 ns MD simulation of in silico generated structure of the mutant. The structure equilibrated (in two separate runs) in similar pattern as the wild type (Fig. S2, Table S3†). In the trajectory of the W113A the residues in the vicinity of the W113A mutation site showed higher RMSFs than in the WT FC (Fig. S3†). Importantly RMSFs of W113A also showed sensitive increase in fluctuations of α2, α3 and α4 (residue 105–145) α-helices, which are involved in the dimerization of WTFC (Fig. 3).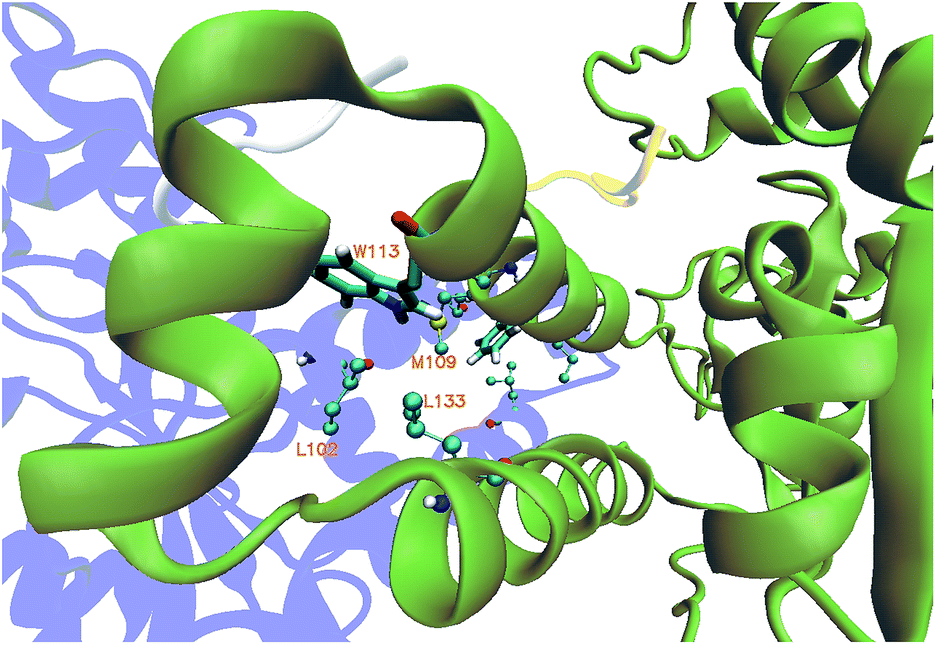 | ||
| Fig. 2 The hydrophobic interactions mediated by residue L102, L133 and M109 in wild type TPST-2 obtained from cluster analysis of WTFC for 100 ns trajectory. The protomer A and B are represented in blue and green colour in new cartoon representation and the W113 is shown in licorice representation using VMD. | ||
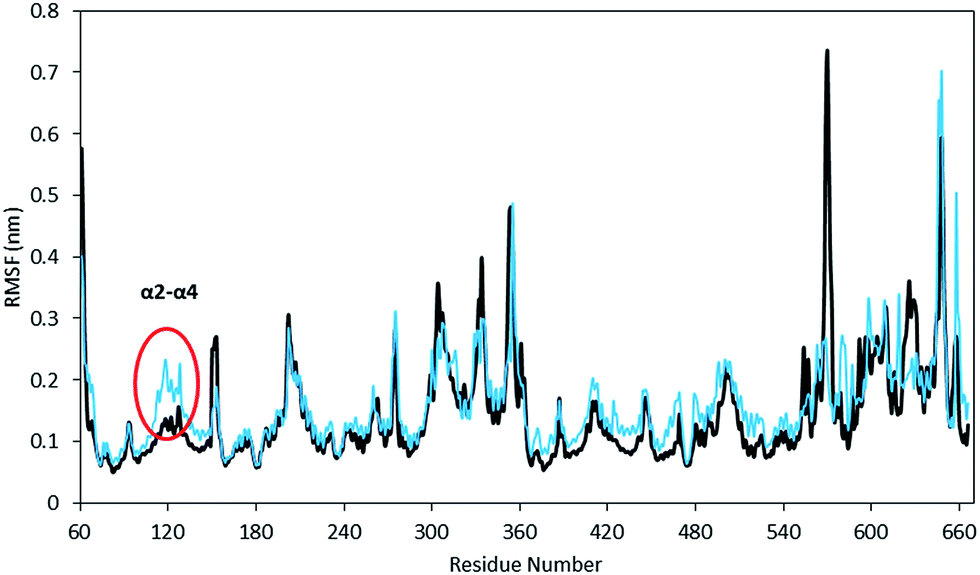 | ||
| Fig. 3 The Root Mean Square Fluctuations (RMSF) of mutant W113A (cyan) with respect to wild type TPST-2 (black) for 100 ns trajectory using Cα atoms. The region α2–α4 (marked with red circle) involved in dimerization shows increase fluctuation in W113A. | ||
In the W113A mutant the distance between the A113 side chain and the side chains of R110, R105, M109 and L133 is higher than that in the WTFC in the MD trajectory (Fig. S5 and Table S2†). This difference indicates about the roles of the above interactions in the formation of the active dimer. The dimerization is very important for the formation of the substrate binding region in WTFC.12 The RMSFs of the substrate itself and the substrate binding region in the W113A mutant are higher than in the WTFC (Fig. 4). These findings indicate the importance of the dimer for the proper formation and the stability of the substrate binding site and are in agreement with the experiments showing that the W113A mutant has reduced the enzyme activity 75%, as compare to the wild type.11
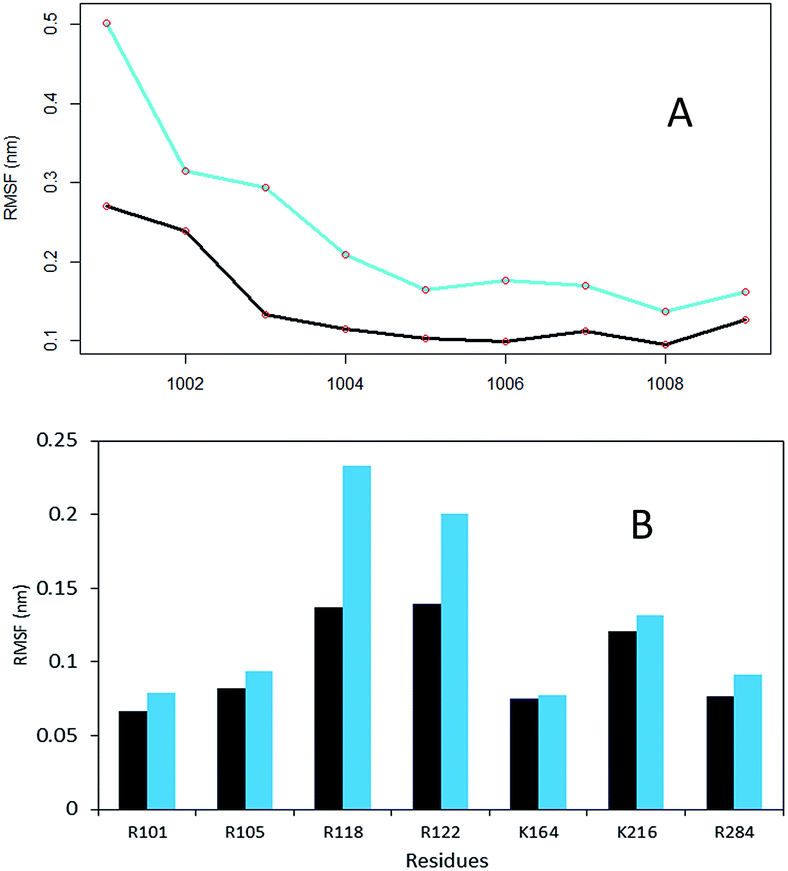 | ||
| Fig. 4 The RMSF of substrate peptide and the substrate binding region of wild type TPST-2 in comparison to W113A mutant for 100 ns trajectory using Cα atoms. (A) The RMSF of the substrate peptide in WTFC (black) and mutant W113A (cyan). (B) The substrate binding residues comparison in WTFC (black) and mutant W113A (cyan). | ||
A single mutation might influence not only the very local structure, surrounding the site of the mutation, but also could have a more global effect on the protein structure and interactions. In the context of dynamics a single mutation might influence the local flexibility around the site, but would also have sensitive impact on the correlated motions between different parts of the protein molecule.
Unique insight about the correlations between movements of different parts of the protein can be provided by the dynamic cross-correlation analysis (DCCA). In order to evaluate the effect of the W113A mutation on the correlated motions in TPST-2, we performed DCCA of the W113A mutant and compared it to the pattern of motions in WTFC. Importantly the DCCA reveals that in the mutant, W113A, there are more anti correlated regions than in the WT FC (Fig. 5). In addition in W113A mutant, the region (α2, α3 and α4) shows reduced correlation with respect to α2, α3 and α4 of WTFC. These residues are involved in dimerization (Fig. 4B). Moreover, residues 180–210 (3′-phosphate binding motif and βe i.e. substrate binding region) of protomer A show anticorrelation towards residues (α2, α3 and α4) of protomer B, which explains the role of W113 in the dimerization and is in agreement with the experimental data about this mutant.
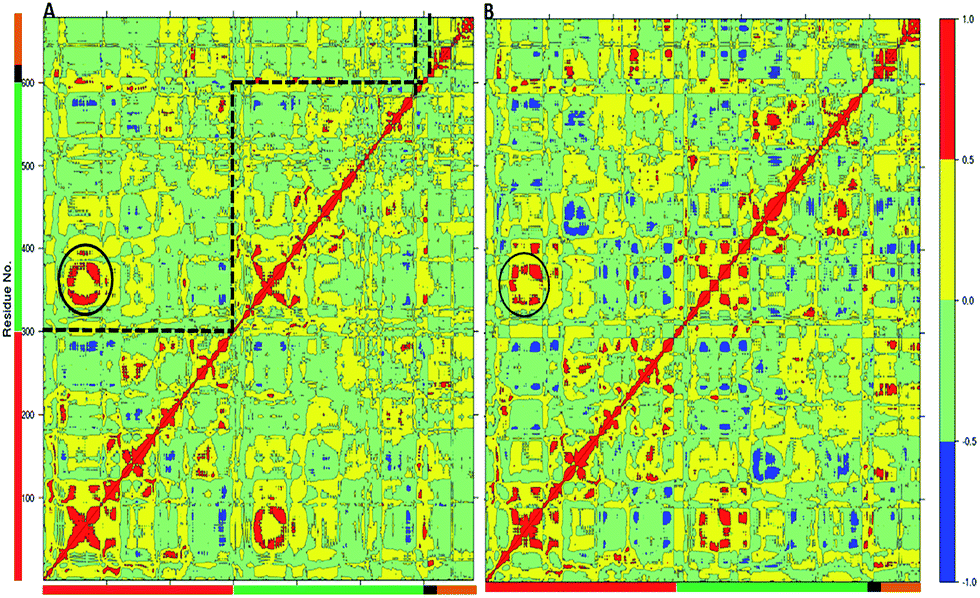 | ||
| Fig. 5 The Dynamic Cross Correlation Analysis (DCCA) of wild type TPST-2 and W113A dimerization mutant. (A) DCCA of wild type TPST-2 as in ref. 16, (B) DCCA of W113A for 100 ns trajectory. The α2–α4 are represented by circular region in black colour. | ||
The MD analysis of the TPST-2 dimer complements the experimental crystallographic and kinetic studies, providing information about new hydrophobic interactions which are not observed in the crystal structure. In addition the simulations of the W113A dimerization, provide structural explanation of its functional effect, namely the mutation leads to increase of distances to important residues, involved in the dimer stabilization, increase in the flexibility of the substrate and the substrate binding site and enhanced flexibility of the alpha-helices (α2–α4) involved in the dimer stabilization.
Conformational flexibility and correlated motions upon binding of the cofactor and the substrate
The MD study of the whole complex, containing the protein dimer, the substrate and the cofactor (WTFC)16 has revealed the key importance of the conformational flexibility which cannot be understood by experimental methods. However, it is crucially important to understand the atomistic nature of the structural changes resulting from the binding of each ligand – PAPS and C4P5Y3. As far as the only available crystal structure so far is the one of the full complex (WTFC),12 MD simulations can provide the needed structural insight as fast alternative of the experimentally determined structures.31 More importantly, an MD analysis of each of the APO, EC and ES in reference to the WTFC would provide unique knowledge about important changes in the correlated motions upon the binding of the substrate and the cofactor which cannot be obtained experimentally.In order to provide these insights into the conformational changes associated with binding of the cofactor PAPS and the substrate peptide C4P5Y3, we simulated for 100 ns: (i) the apoenzyme (APO) built from protomers A and B; (ii) the apoenzyme plus the cofactor PAPS complex (EC); (iii) the apoenzyme plus the substrate C4P5Y3 complex (ES) and (iv) the substrate in water (S) and compared them with the MD simulations of WT FC.12
| Structure | RMSD (Å) | RMSF > 1 Å (%) | RMSF < 1 Å (%) | |
|---|---|---|---|---|
| Mean (μ) | SD (σ2) | |||
| WTFC | 2.72 | 0.27 | 57 | 43 |
| Apoenzyme | 4.37 | 0.57 | 79 | 21 |
| Enz. + substr. | 3.32 | 0.35 | 65 | 35 |
| Enz. + cofact. | 4.52 | 0.52 | 75 | 25 |
| Protomer A | 3.98 | 0.67 | 66 | 34 |
| Substrate | 3.28 | 0.56 | 100 | 0 |
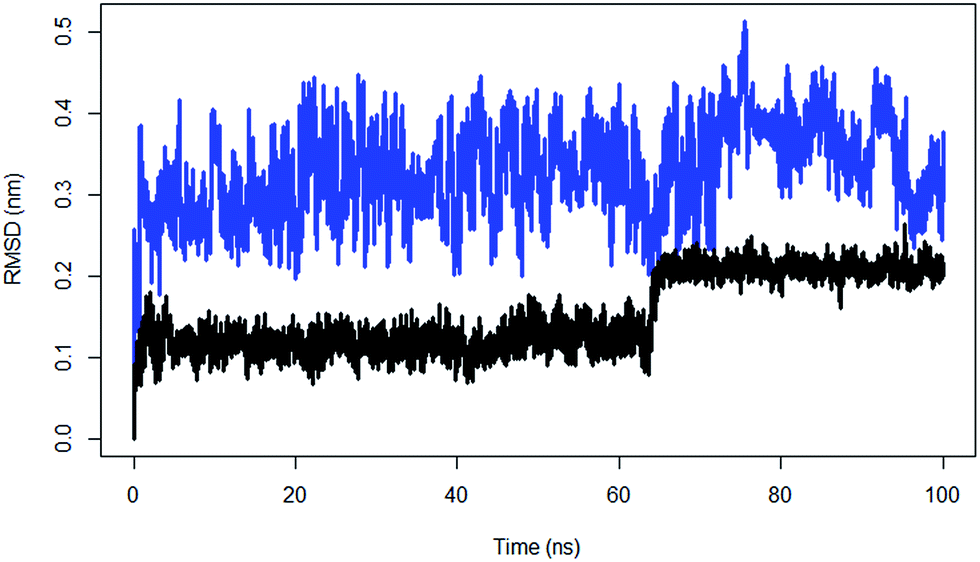 | ||
| Fig. 6 The RMSD of substrate peptide of the wild type TPST-2 (black) compared against substrate peptide only in water (blue) for 100 ns using Cα atoms. | ||
The ES complex showed increased fluctuations in the 5′PBS (5′-phosphosulfate binding motif) region, which indicates that the binding of PAPS has stabilizing effect on the 5′PBS. The EC shows an increase in fluctuation of R78, S285 and some part of the 3′PBS (3′-phosphate binding motif) region thus suggesting that substrate binding has a stabilizing effect for the cofactor and substrate binding sites in WTFC (Fig. S6†). The average value of radius of gyration (Rg) of WTFC is 28.7 Å, the Rg of EC increased to 30.2 Å and for ES is 28.8 Å (Table S4, Fig. S7†) demonstrating the effect of the substrate binding on compression of the structure of the full complex. These results show that the binding of both the substrate and the cofactor contribute for the stability of the full active complex; however, the effect of the substrate binding is much more prominent. In the EC complex R78 makes a stable interaction with the 5′PBS region of the cofactor as in WT FC (Fig. S8†). The S285 hydroxyl group is hydrogen bonded to OAF residue of 5′PBS site during only 54% of the simulation. The hydroxyl group of S285, makes interactions with the OAD oxygen of the 3′ phosphate group in contrast to the WTFC where it interacts with the OAF of 5′PBS region. The side chain of R183 in WTFC stabilizes the 3′PBS phosphate OAB oxygen atom (average distance 3.63 Å) however in EC complex it makes a weaker interaction (average distance 5.1 Å). The RMSF of PAPS in the EC complex shows higher fluctuations compared to WTFC (Fig. S9†) which indicates the stabilizing role of the substrate for PAPS cofactor. The changes in the above atomistic interactions illustrate the key structural changes which occur upon binding of the substrate and the cofactor and reinforce the idea of accounting for the conformational flexibility whilst modelling protein–ligand interactions (e.g. in the docking studies).
The current MD study reveals the importance of conformational flexibility for the dimerization of TPST-2. Enzymes often exist as dimers in physiological conditions and carry out their biochemical functions in such state; therefore computational modelling of the dimer state provides important insight about their structure–function relationships which can be used for further modelling of their reaction mechanisms, (e.g. as in the case of the multiscale modelling of the COX-1 enzyme34).
Our results support the idea that the conformational changes associated with interactions with ligands (substrates, cofactors, inhibitors) can lead to changes of the system of interactions within the entire enzyme molecule, rather than only in the local binding site. In similar way for example, the network of interactions between the aromatic chromophores, which generate the near-UV circular dichroism in class-A β-lactamases and human carbonic anhydrase are sensitively influenced by the conformational changes, related to the binding of ligands.13,35–37 The MD studies presented here assert about the importance of accounting for the conformational flexibility in computational docking studies of protein–ligand interactions.
Conclusions
The present MD study reveals the key effect of the protein flexibility on the dimerization of TPST-2 and the interactions between the apoenzyme, the cofactor PAPS and the substrate peptide C4P5Y3. New hydrophobic interactions, important for the protomer–protomer interactions are identified and the structural effect of the key dimerization mutation W113A is explained in the absence of experimentally-derived structure of the mutant. Our results show that the binding of the substrate and cofactor influence not only the local interactions in the binding site, but also affect the correlated motions in the entire molecule. The study asserts about the importance of accounting for conformational dynamics in studying protein–ligand and protein–protein interactions.Author contributions
W. S. wrote the manuscript and carried out molecular dynamics simulations and analyzed the results. C. C. and T. K. designed the experiment; analyzed and discussed results wrote and revised manuscript. G. W., O. S. and P. P. discussed the results and revised manuscript.Acknowledgements
TK and CC acknowledge Marie Curie International Career Development Fellowships, NSCCS grants and HEC-Biosim grants. WS thanks for Northumbria University Studentship. The authors acknowledge the departmental HPC-Cluster “Pasteur”.Notes and references
- C. Niehrs, J. Stinchcombe and W. Huttner, Two membrane-bound forms of tyrosylprotein sulfotransferase as revealed by phase partitioning in Triton X-114, Eur. J. Cell Biol., 1992, 58, 35–43 CAS.
- C. Niehrs and W. B. Huttner, Purification and characterization of tyrosylprotein sulfotransferase, EMBO J., 1990, 9, 35 CAS.
- J. W. Kehoe and C. R. Bertozzi, Tyrosine sulfation: a modulator of extracellular protein–protein interactions, Chem. Biol., 2000, 7, R57–R61 CrossRef CAS PubMed.
- M. J. Stone, S. Chuang, X. Hou, M. Shoham and J. Z. Zhu, Tyrosine sulfation: an increasingly recognised post-translational modification of secreted proteins, New Biotechnol., 2009, 25, 299–317 CrossRef CAS.
- H. Choe and M. Farzan, Chapter 7: tyrosine sulfation of HIV-1 coreceptors and other chemokine receptors, Methods Enzymol., 2009, 461, 147–170 CAS.
- Y. Nishimura, T. Wakita and H. Shimizu, Tyrosine sulfation of the amino terminus of PSGL-1 is critical for enterovirus 71 infection, PLoS Pathog., 2010, 6, e1001174 Search PubMed.
- S. Kraemer, S. Alampour-Rajabi, O. E. Bounkari and J. Bernhagen, Hetero-oligomerization of chemokine receptors: diversity and relevance for function, Curr. Med. Chem., 2013, 20, 2524–2536 CrossRef CAS PubMed.
- L. Zhu, Q. Zhao and B. Wu, Structure-based studies of chemokine receptors, Curr. Opin. Struct. Biol., 2013, 23, 539–546 CrossRef CAS PubMed.
- R. Beisswanger, D. Corbeil, C. Vannier, C. Thiele, U. Dohrmann, R. Kellner, K. Ashman, C. Niehrs and W. B. Huttner, Existence of distinct tyrosylprotein sulfotransferase genes: molecular characterization of tyrosylprotein sulfotransferase-2, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11134–11139 CrossRef CAS.
- Y.-B. Ouyang and K. L. Moore, Molecular cloning and expression of human and mouse tyrosylprotein sulfotransferase-2 and a tyrosylprotein sulfotransferase homologue in Caenorhabditis elegans, J. Biol. Chem., 1998, 273, 24770–24774 CrossRef CAS PubMed.
- T. Teramoto, Y. Fujikawa, Y. Kawaguchi, K. Kurogi, M. Soejima, R. Adachi, Y. Nakanishi, E. Mishiro-Sato, M.-C. Liu and Y. Sakakibara, Crystal structure of human tyrosylprotein sulfotransferase-2 reveals the mechanism of protein tyrosine sulfation reaction, Nat. Commun., 2013, 4, 1572 CrossRef PubMed.
- T. Teramoto, Y. Fujikawa, Y. Kawaguchi, K. Kurogi, M. Soejima, R. Adachi, Y. Nakanishi, E. Mishiro-Sato, M.-C. Liu and Y. Sakakibara, Crystal structure of human tyrosylprotein sulfotransferase-2 reveals the mechanism of protein tyrosine sulfation reaction, Nat. Commun., 2013, 4, 1572 CrossRef PubMed.
- T. G. Karabencheva-Christova, U. Carlsson, K. Balali-Mood, G. W. Black and C. Z. Christov, Conformational effects on the circular dichroism of human carbonic anhydrase II: a multilevel computational study, PLoS One, 2013, 8, e56874 CAS.
- M. Karplus and J. A. McCammon, Molecular dynamics simulations of biomolecules, Nat. Struct. Biol., 2002, 9, 646–652 CrossRef CAS PubMed.
- S. A. Adcock and J. A. McCammon, Molecular dynamics: survey of methods for simulating the activity of proteins, Chem. Rev., 2006, 106, 1589–1615 CrossRef CAS PubMed.
- W. Singh, T. G. Karabencheva-Christova, G. W. Black, O. Sparagano and C. Z. Christov, Conformational flexibility influences structure–function relationships in tyrosyl protein sulfotransferase-2, RSC Adv., 2016, 6, 11344–11352 RSC.
- W. Singh, T. G. Karabencheva-Christova, G. W. Black, J. Ainsley, L. Dover and C. Z. Christov, Conformational Dynamics, Ligand Binding and Effects of Mutations in NirE an S-Adenosyl-L-Methionine Dependent Methyltransferase, Sci. Rep., 2016, 6, 20107 CrossRef CAS PubMed.
- T. G. Karabencheva, C. C. Lee, G. W. Black, R. Donev and C. Z. Christov, How does conformational flexibility influence key structural features involved in activation of anaplastic lymphoma kinase?, Mol. BioSyst., 2014, 10, 1490–1495 RSC.
- S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- H. J. Berendsen, D. van der Spoel and R. van Drunen, GROMACS: a message-passing parallel molecular dynamics implementation, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- D. van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, GROMACS: fast, flexible, and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- L. D. Schuler, X. Daura and W. F. van Gunsteren, An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase, J. Comput. Chem., 2001, 22, 1205–1218 CrossRef CAS.
- R. Fletcher and M. J. Powell, A rapidly convergent descent method for minimization, Comput. J., 1963, 6, 163–168 CrossRef.
- M. R. Hestenes and E. Stiefel, Methods of conjugate gradients for solving linear systems, 1952 Search PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, Interaction models for water in relation to protein hydration, Intermolecular forces, ed. B. Pullman, 1981, pp. 331–342 Search PubMed.
- I. R. McDonald, NpT-ensemble Monte Carlo calculations for binary liquid mixtures, Mol. Phys., 1972, 23, 41–58 CrossRef CAS.
- P. P. Ewald, Die Berechnung optischer und elektrostatischer Gitterpotentiale, Ann. Phys., 1921, 369, 253–287 CrossRef.
- B. Hess, P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation, J. Chem. Theory Comput., 2007, 4, 116–122 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed , 27-8.
- B. J. Grant, A. P. Rodrigues, K. M. ElSawy, J. A. McCammon and L. S. Caves, Bio3d: an R package for the comparative analysis of protein structures, Bioinformatics, 2006, 22, 2695–2696 CrossRef CAS PubMed.
- D. Kruschel and B. Zagrovic, Conformational averaging in structural biology: issues, challenges and computational solutions, Mol. BioSyst., 2009, 5, 1606–1616 RSC.
- K. Kasahara, I. Fukuda and H. Nakamura, A Novel Approach of Dynamic Cross Correlation Analysis on Molecular Dynamics Simulations and Its Application to Ets1 Dimer–DNA Complex, 2014 Search PubMed.
- B. L. Kormos, A. M. Baranger and D. L. Beveridge, Do collective atomic fluctuations account for cooperative effects? Molecular dynamics studies of the U1A–RNA complex, J. Am. Chem. Soc., 2006, 128, 8992–8993 CrossRef CAS PubMed.
- C. Christov, A. Lodola, T. Karabencheva, S. Wan, P. Coveney and A. Mulholland, Conformational Effects on the pro-S Hydrogen Abstraction Reaction in Cyclooxygenase-1: An Integrated QM/MM and MD Study, Biophys. J., 2013, 104, L5–L7 CrossRef CAS PubMed.
- C. Christov and T. Karabencheva, Mechanisms of generation of rotational strengths of Class-A β-lactamase from Escherichia coli (TEM-1) part I: theoretical analysis of the influence of conformational changes in the near UV, Chem. Phys. Lett., 2004, 396, 282–287 CrossRef CAS.
- C. Christov and T. Karabencheva, Computational Insight in Protein Circular Dichroism: Detailed Analysis of Contributions of Individual Chromophores in TEM-1 β-Lactamase, Theor. Chem. Acc., 2010, 128, 25–37 CrossRef.
- C. Christov, T. Karabencheva, A. Lodola., Relationship between chiroptical properties, conformational changes and interactions in.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01899h |
| This journal is © The Royal Society of Chemistry 2016 |