
Two new mixed copper(ii)–dipeptide complexes of N,N-donor heterocycle ligands: studies on their non-covalent DNA binding, chemical nuclease, antioxidant and anticancer activities
Abstract
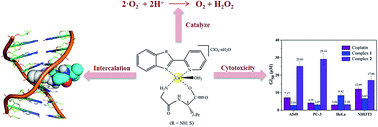
Two novel mononuclear mixed ligand copper(II) complexes, [Cu(Gly-L-val)(HPB)(H2O)]·ClO4·1.5H2O (1) and [Cu(Gly-L-val)(PBT)(H2O)]·ClO4 (2) (Gly-L-val = glycyl-L-valine, HPB = 2-(2′-pyridyl)benzimidazole, PBT = 2-(2′-pyridyl)benzothiazole), have been synthesized and characterized using various analytical and spectroscopic methods. The interactions of the complexes with DNA have been explored by viscometry, thermal denaturation, cyclic voltammetry (CV), agarose gel electrophoresis and spectroscopic means (UV absorption, circular dichroism (CD) and fluorimetry), as well as molecular docking techniques. These studies confirmed the mode of the complexes bound to calf thymus DNA (CT-DNA) through insertion with certain affinity (Kb = 3.211 × 105 M−1 for 1 and 4.734 × 104 M−1 for 2). In the fluorimetric experiments of thermodynamics (KSV = 1.145 × 104 M−1 for 1 and 2.634 × 103 M−1 for 2), the changes in enthalpy (ΔH > 0), entropy (ΔS > 0) and Gibbs free energy (ΔG < 0) in the interactions between the complexes with DNA suggested that the process occurred spontaneously through hydrophobic interactions. The complexes displayed oxidative cleavage of pBR322 plasmid DNA in the presence of ascorbic acid, probably induced by ˙OH as a reactive oxygen species. Furthermore, the molecular docking technique was applied to ascertain the mode of action for the complexes towards DNA. Moreover, superoxide dismutase (SOD) activity studies were performed using the photoreduction of nitroblue tetrazolium (NBT) under a non-enzymatic system and the antioxidant activities of 1 and 2 determined with IC50 values of 0.337 and 0.146 μM, respectively. The cytotoxicity of the Cu(II) complexes against A549, HeLa, PC-3 tumor cell lines and NIH3T3 (non-tumor cell line) was studied by an MTT assay and it was found that 1 exhibited better cytotoxicity against A549 and PC-3 than 2 and the widely used drug cisplatin.
 Please wait while we load your content...
Please wait while we load your content...

