
A catch–release catalysis system based on supramolecular host–guest interactions†
Abstract
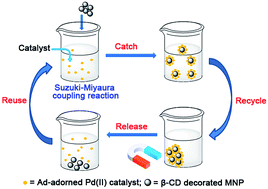
A generic catch–release catalysis system has been designed for the recovery of homogeneous catalysts at the end of a reaction using host–guest interactions. This proof-of-concept system consists of a palladium(II)–dipyrazole complex bearing an adamantyl (Ad) molecular recognition moiety (guest), and magnetic nanoparticles (MNP) decorated with β-cyclodextrins (β-CD) (host). The density of β-CD on the iron oxide nanoparticles was up to 3.1 × 10−4 mmol per mg, sufficient to efficiently catch the Ad-adorned Pd catalyst in aqueous methanol at room temperature. Release and recycling of the catalyst was achieved by extraction with methanol. This catch–release system performed well in the Suzuki–Miyaura coupling, but suffers from slow degradation which restricts the number of times that the catalyst and magnetic nanoparticles can be recycled and reused.
 Please wait while we load your content...
Please wait while we load your content...

