
The redispersion behaviour of Pt on the surface of Fe2O3†
Jingwei Liab,
Yibo Zhang*a,
Ting Yiab,
Zeshu Zhangab,
Zhenzhen Miaoa,
Liwei Suna,
Zhendong Zhanga and
Xiangguang Yang*a
aState Key Laboratory of Rare Earth Resource Utilization, Green Chemistry and Process Laboratory, Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences, Changchun, Jilin 130022, China. E-mail: xgyang@ciac.ac.cn; yibozhang@ciac.ac.cn; Tel: +86-431-8526-2228
bUniversity of Chinese Academy of Sciences, Beijing 100039, China
First published on 26th February 2016
Abstract
The redispersion behaviour of Pt on ferric oxides under cyclic oxidizing/reducing conditions was observed and studied using several analysis techniques. Pt in a Pt/Fe2O3 catalyst sintered after treatment under a single oxidative or reductive atmosphere at 600 °C and the average size of the Pt nanoparticles increased from the original 3.2 nm to 6.8 and 14.6 nm, respectively. In contrast, the average size decreased to 1.1 nm when the catalyst was treated under an alternating oxidizing and reducing atmosphere. It is proposed that the Pt–O–Fe bond formed under an oxidative atmosphere and the related defects or oxygen vacancies formed under a reductive atmosphere play an important role during the redispersion of Pt. In addition, the effect of temperature on the redispersion of Pt is also noted and discussed.
Introduction
Among commercial catalysts, supported noble metal catalysts (e.g., platinum, palladium and rhodium) are widely utilized in various industrial processes,1–3 such as the removal of volatile organic compounds (VOCs),4,5 the selective hydrogenation or oxidation of hydrocarbons6–8 and the production of hydrogen.9,10 In particular, supported Pt catalysts have attracted continuous attention because of their marvelous catalytic performance in emission control/treatment11 and the refining of fuel stock.12–14 Based on the fact that Pt is fairly expensive and difficult to replace with other components, one of the biggest issues relating to supported Pt catalysts is how to prolong the life-time of the catalysts or regenerate the deactivated ones effectively.Generally, highly dispersed noble metal nanoparticles are more active than bulk ones for the majority of catalytic reactions. However, they often tend to agglomerate into larger particles, especially at higher reaction temperatures, leading to the degradation of catalytic activity and even a loss of the unique catalytic properties observed in the original nanoparticles. The redispersion of noble metals is an effective method to reverse sintering during reactions or to regenerate spent catalysts for reuse.15–18 In the cases of Rh and Pd, they can be efficiently redispersed simply using flowing oxygen at certain temperatures.19 Nevertheless, the redispersion of Pt might be not that easy. For instance, the common method for the redispersion of noble metals mentioned above is not suitable for Pt because even when treated at the optimal operating conditions (around 500 °C), the redispersion of Pt is still at a low level.20,21 As for other methods, the conditions are comparatively rigorous, for example the support is restricted to substrates containing ceria22,23 or corrosive chlorine must be introduced during treatment.24
Herein, we find that Pt can be redispersed effectively on the surface of Fe2O3 through alternating treatments under oxidative and reductive atmospheres. When treated at 600 °C, most Pt nanoparticles with an average size of 3.2 nm were redispersed to 1.1 nm. In addition, CO oxidation was used as a probe reaction to explore the catalytic performance of the Pt/Fe2O3 samples before and after redispersion. As we know, it is an expected trend that supported noble metal catalysts with smaller particle sizes (within a certain limit) are beneficial to the enhancement of catalytic activity, especially for simple catalytic reactions.15–18 The results of the activity test showed that the catalytic activity of Pt/Fe2O3 was obviously enhanced with the decrease in the size of the Pt nanoparticles.
Experimental
Chemicals
All the reagents were used as received without further purification. H2PtCl6·6H2O was obtained from Shanghai Shi Wu Chemical Reagent Science and Technology Co. Ltd. Polyvinylpyrrolidone (PVP) and FeCl3·6H2O were obtained from Aladdin Chemicals Co. Ltd.Catalyst preparation
Pt/Fe2O3 was prepared through the wet impregnation of Fe2O3 with a Pt sol. A slurry of Pt NPs and Fe2O3 was stirred for 1 h at room temperature and sonicated for 30 min. Then, the mixture was evaporated at 60 °C with stirring and calcined at 400 °C for 3 h.
The Pt/Fe2O3 catalyst was also prepared through a conventional impregnation method: an appropriate amount of H2PtCl6 solution (0.03 M) was added to the Fe2O3 powder and the mixture was evaporated to dryness at 60 °C under stirring to obtain 3.1 wt% Pt/Fe2O3. Then the catalyst was dried at 110 °C for 10 h, calcined at 450 °C for 3 h and reduced in a H2 flow at 200 °C for 2 h. The synthesized catalyst was denoted as Pt/Fe2O3(IM).
Characterization
The CO oxidation activities of the catalysts were measured using a fixed-bed flow reactor. 25 mg of sample was loaded into a quartz tube and supported using quartz wool. The reactant gas (1% CO/air vol%) was passed through the catalytic bed at a flow rate of 25 mL min−1 and measured using a gas chromatograph equipped with a thermal conductivity detector.The samples were treated in a system with a switch valve and temperature controlling system (Fig. 1) which allowed exposure to the various atmospheres under investigation. 25 mg of sample was heated to 500–700 °C at a heating rate of 10 °C min−1 under a certain atmosphere (20% O2/N2, 4% H2/Ar or an alternating atmosphere of these two kinds of gases) at a flow rate of 100 mL min−1 and maintained at that temperature for 30 min, then cooled down to room temperature under the same atmosphere. In the redispersion experiment, samples were treated under an alternating atmosphere of 20% O2/N2 for 5 seconds and 4% H2/Ar for 10 seconds. 10% H2O vapor was produced by bubbling Ar through water at 46 °C at 0.1 MPa saturated vapor pressure. The samples treated in 20% O2/N2, 4% H2/Ar and alternating gases of 20% O2/N2 and 4% H2/Ar at different temperatures were denoted as Pt/Fe2O3(600 O), Pt/Fe2O3(600 H) and Pt/Fe2O3(500 H–O, 600 H–O or 700 H–O), respectively.
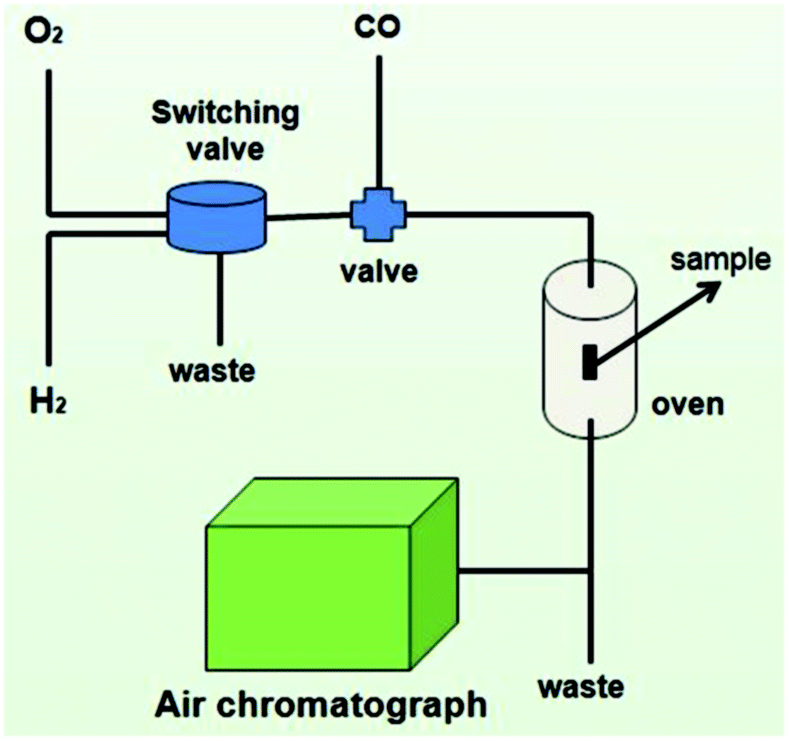 | ||
| Fig. 1 Sketch map of the system used for treating samples under various atmospheres and CO oxidation. | ||
The powder X-ray diffraction (XRD) patterns were obtained with a Bruker D8 Advance X-ray diffractometer using a Cu Kα radiation source (λ = 1.5406 Å). The powder samples were placed on a glass slide and scanned from 10° to 90° at a rate of 3° min−1. The elemental content was measured using ICP-OES (iCAP 6300 Thermo Scientific USA). The hydrogen temperature-programmed reduction (H2-TPR) experiments were performed on a Micromeritics AutoChem 2920 instrument. Before H2-TPR, the samples were heated at 400 °C in nitrogen for 30 min and then cooled to room temperature. The samples were reduced in a 10% H2/Ar flow with a heating rate of 10 °C min−1. Surface morphological images were obtained using scanning electron microscopy (SEM) (Hitachi S-4800) after gold plating at an accelerating voltage of 10 kV. Transmission electron microscopy (TEM) was performed using an FEI Tecnai G2 at an accelerating voltage of 200 kV. X-Ray Photoelectron Spectroscopy (XPS) was performed using a VG Thermo ESCALAB 250 spectrometer operated at 120 W.
Results and discussion
TEM analyses
The PVP-protected Pt NPs are dispersed uniformly with an average particle size of 3.2 nm (Fig. 2a). In the case of the FeOOH NRs (the precursor of the Fe2O3 NRs), each nanorod exhibits an average width of ca. 50 nm and a length of ca. 300 nm (Fig. 2b). As indicated in Fig. 2c, the FeOOH NRs convert into hollow Fe2O3 NRs after calcination. The Pt NPs are well dispersed and remain unchanged after loading on the Fe2O3 NRs (Fig. 2d). The loading content of Pt is 3.1 wt%, as determined using ICP analysis. | ||
| Fig. 2 TEM images of Pt NPs (a), FeOOH NRs (b), Fe2O3 NRs (c) and Pt/Fe2O3 (d). The inset in (a) shows the statistical Pt NP distribution and N indicates the total statistical number of Pt NPs. | ||
As shown in Fig. 3a and b, growth of the Pt NPs is observed when the Pt/Fe2O3 samples are treated in 20% O2/N2 or 4% H2/Ar. Concretely, the average size of the Pt NPs increases to 6.8 nm under an oxidative atmosphere and 14.6 nm under a reductive atmosphere. Meanwhile, the support Fe2O3 is destroyed to some extent, especially under a reductive atmosphere. In sharp contrast, a large amount of Pt NPs with small sizes appear after H–O treatment (Fig. 3c–h). The average sizes of the Pt NPs are 2.5, 1.1 and 2.8 nm after H–O treatment at 500, 600 and 700 °C, respectively. Particularly, few large Pt NPs could be observed in the Pt/Fe2O3(600 H–O) sample and the proportion of Pt NPs with sizes smaller than 1.5 nm reaches 80%. As for the fresh Pt/Fe2O3(IM) sample, the sizes of the Pt NPs are not uniform and range from 1 to 9 nm (Fig. S1†). After H–O treatment at 600 °C, most Pt NPs are around 2 nm in diameter, indicating that the redispersion behaviour of Pt on Fe2O3 applies to the common catalyst prepared through the impregnation method as well.
 | ||
| Fig. 3 TEM images of Pt/Fe2O3 samples after treatment under various conditions. (a) In 20% O2/N2 at 600 °C; (b) in 4% H2/Ar at 600 °C; (c, d) in alternating H–O at 500 °C; (e, f), in alternating H–O at 600 °C; (g, h), in alternating H–O at 700 °C. | ||
Catalytic performance for CO oxidation
Fig. 4 exhibits the catalytic activities of fresh Pt/Fe2O3 and Pt/Fe2O3 after different treatments. Compared to the fresh Pt/Fe2O3 catalyst, which could completely oxidize CO at 210 °C, the catalytic activities of catalysts after H–O treatment at different temperatures are improved to different degrees, and the temperatures required for complete conversion of CO (TCC) are 190 (500 H–O), 150 (600 H–O) and 180 °C (700 H–O). However, a reverse trend occurs for Pt/Fe2O3(600 O) and Pt/Fe2O3(600 H) and the TCC are 260 and 450 °C, respectively. The redispersion of Pt on the surface of Fe2O3 results in the production of a large amount of Pt NPs with smaller sizes, which means that there are more active centres for CO oxidation and the size effect of Pt is more significant. | ||
| Fig. 4 CO conversion as a function of reaction temperature for Pt/Fe2O3 catalysts before and after various treatments. | ||
Redox properties
H2-TPR is an effective method to test the reducibility of samples. For a fresh Fe2O3 sample (as shown in Fig. 5), the reduction peaks located around 415 and 600 °C are attributed to the reduction of Fe2O3 → Fe3O4 and Fe3O4 → FeO → Fe.25 After the loading of Pt, the reduction temperatures of these two peaks shift to lower temperature ranges, indicating that the presence of Pt promotes the reduction of Fe2O3 significantly. A very weak reduction peak at about 140 °C is observed, which is primarily attributed to the reduction of Pt2+ to Pt0, along with some contribution from the partial reduction of iron oxide linked with the Pt species directly (i.e., Fe–O–Pt).26 Additionally, the peak at 220 °C is assigned to the reduction of oxidized Pt species (e.g., Pt4+ to Pt0),26 which partially overlaps with the reduction of Fe2O3 to Fe3O4 (at 270 °C). In the case of the Pt/Fe2O3 samples after H–O treatment, the low-temperature reduction peak centred at 140 °C disappears. Furthermore, the two reduction peaks (220 and 270 °C) integrate into a single one, the hydrogen consumption of which is remarkably less than the sum of the former two peaks, suggesting that the oxidation states of the Pt species decrease after H–O treatment. It should be noted that the single reduction peak shifts to a lower temperature region for Pt/Fe2O3(600 H–O), which is due to the fact that the small Pt NPs formed during H–O treatment at 600 °C could improve the spillover of H atoms from the Pt species to Fe2O3 more effectively.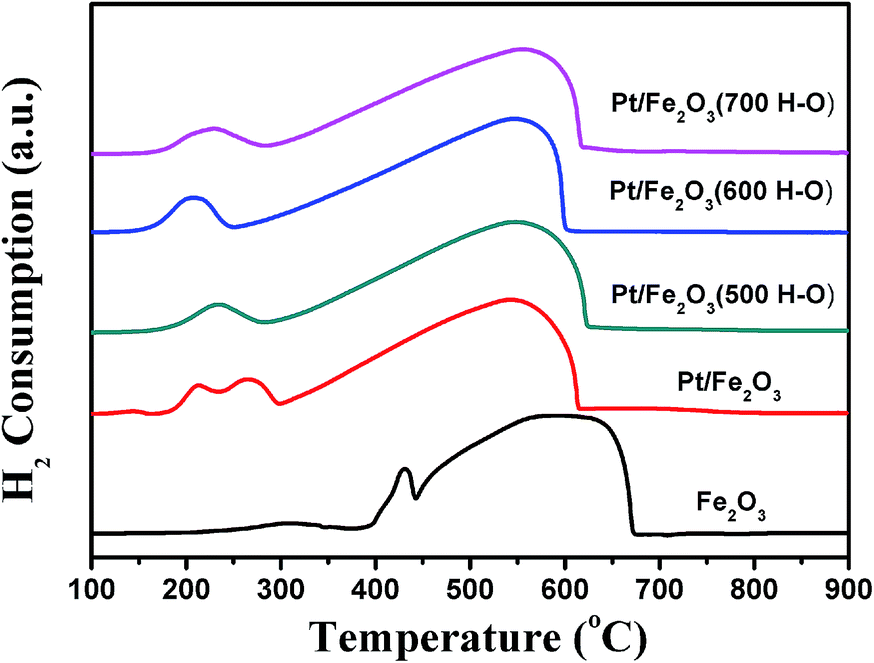 | ||
| Fig. 5 H2-TPR profiles of Fe2O3, Pt/Fe2O3 and Pt/Fe2O3 after H–O treatment at different temperatures. | ||
XRD characterization
Wide angle XRD patterns for FeOOH, Fe2O3, Pt/Fe2O3 and Pt/Fe2O3 after H–O treatment are shown in Fig. 6. For the Fe2O3 and Pt/Fe2O3 samples, only peaks ascribed to α-Fe2O3 (PDF#33-0664) are observed and no signals corresponding to Pt species are detected. This is presumably attributed to the fact that the Pt NPs in the measured sample are not large enough to generate a detectable signal or the diffraction peaks from Fe2O3 have covered the weak ones of Pt. Moreover, in the case of the Pt/Fe2O3 samples after H–O treatment, some new peaks located at approximately 30.4°, 43.3°, 53.6° and 57.2° appear, which are ascribed to γ-Fe2O3 (further confirmed by XPS analyses later), and the partial magnification patterns are presented in Fig. S2.† It is well known that the crystal phase transformation between α-Fe2O3 and γ-Fe2O3 could occur easily under reductive and oxidative atmospheres in the range from 400–600 °C.27,28 The results suggest that on the surface of Fe2O3, continuous crystal phase transformation between α-Fe2O3 and γ-Fe2O3 happens during the H–O treatment, accompanied by the generation of a large amount of relative defects. Compared to Pt/Fe2O3(500 H–O) and Pt/Fe2O3(700 H–O), the diffraction intensity of γ-Fe2O3 for the Pt/Fe2O3(600 H–O) sample is much higher, indicating that more defects are generated when treated at 600 °C. Furthermore, as we know, the transformation between γ-Fe2O3 and Fe3O4 is quite easy under the experimental conditions due to their similar structure (i.e., Fe3O4 can transform to γ-Fe2O3 slowly in air, even at room temperature).29–33 Both γ-Fe2O3 and Fe3O4 have the same inverse spinel structure, excluding that some oxygen ions in γ-Fe2O3 are substituted with oxygen vacancies in Fe3O4. Namely, the transformation between γ-Fe2O3 and Fe3O4 just refers to the storage and release of O atoms without any change in the basic crystal structure. Therefore, it is reasonable to speculate that the Fe3O4 phase is generated during H–O treatment and transforms to γ-Fe2O3 gradually in the later cooling process. As a result, the Fe3O4 phase disappears completely in the test samples. | ||
| Fig. 6 XRD patterns of FeOOH, Fe2O3, Pt/Fe2O3 and Pt/Fe2O3 after H–O treatment at different temperatures. ● and ♦ represent the diffraction peaks ascribed to α-Fe2O3 and γ-Fe2O3, respectively. | ||
XPS analyses
To gain better insight into the nature of the crystal structure and oxidation states, the Fe 2p and Pt 4f XPS spectra of Pt/Fe2O3 samples before and after H–O treatment are displayed in Fig. 7 (the survey scans are given in Fig. S3†). For fresh Pt/Fe2O3, the XPS spectrum of Fe 2p exhibits two peaks at 710.4 and 723.7 eV,34 which can be attributed to Fe 2p3/2 and Fe 2p1/2, respectively. Furthermore, a satellite peak located at 718.3 eV could also be observed and the binding energy between the satellite and Fe 2p3/2 peak is 7.8 eV. The results above suggest that Fe is in a +3 oxidation state, which is consistent with the XRD tests. As reported in the literature,35 the core level spectra of α-Fe2O3 and γ-Fe2O3 are almost identical. Therefore, the partial transformation from α-Fe2O3 to γ-Fe2O3 would not shift the binding energies significantly. Compared to α-Fe2O3 or γ-Fe2O3, the Fe 2p3/2 and Fe 2p1/2 peaks of Fe3O4 shift to high binding energies and broaden obviously due to the appearance of Fe2+. Moreover, for the Fe 2p peaks of Fe3O4, there is no satellite at about 719 eV.33–35 In our experiments, after H–O treatment, although the binding energy in the Fe 2p spectra shifts to high values, the presence of a satellite peak located at 718.7 eV and the unchanged peak width at half height of Fe 2p indicate that the Fe3O4 phase does not exist in the measured samples and the newly-appearing diffraction peaks for Pt/Fe2O3(500–700 H–O) in the XRD patterns are attributed to γ-Fe2O3. The shift of the Fe 2p peaks could be ascribed to the strong interaction between the support and the small Pt NPs formed during the treatment. As for the spectra of Pt 4f, the peaks shift to a lower binding energy slightly after H–O treatment, suggesting that the oxidation states of Pt decrease, which is consistent with the TPR results. Besides, it is noteworthy that the intensity of Pt/Fe2O3(600 H–O) is lower than the others. It is inferred that the Pt NPs redisperse adequately under these circumstances and some of the small Pt NPs move into a crack in the support during the redispersion process, and cannot be detected using XPS measurements.36 | ||
| Fig. 7 XPS spectra of Fe 2p (a) and Pt 4f (b) for Pt/Fe2O3 samples before and after H–O treatment. | ||
Discussion
On the basis of the above analysis, a possible process for the Pt NPs redispersion on Fe2O3 under alternating atmospheres of oxidation and reduction is proposed as follows: under an oxidative atmosphere, Pt–O–Fe or Pt–O–O–Fe forms on the interface of the Pt NPs and Fe2O3. During this course, the Pt-NPs are flattened to some extent and a part of the Pt atoms on the Pt NPs would be pulled onto the interface; under a reductive atmosphere, PtO is reduced to metallic Pt and a hydrogen spillover from Pt to Fe occurs, followed by the breaking of the Pt–O–Fe or Pt–O–O–Fe bonds. Meanwhile, related defects and oxygen vacancies are formed due to the crystal phase transformation of α-Fe2O3 to γ-Fe2O3 and the partial slight reduction of surface Fe2O3 through hydrogen spillover, respectively. Finally, Pt atoms are trapped at defects and oxygen vacancies. Furthermore, during H–O treatment, the transformation between γ-Fe2O3 and Fe3O4 happens continuously, accompanied by the storage and release of O atoms, just like the “oxygen storage material” ceria. Namely, the crystal transformation between γ-Fe2O3 and Fe3O4 can improve the oxygen mobility of catalysts, which is beneficial for the oxidation of CO and the redispersion of Pt.22In addition, the degrees of redispersion and sintering influenced by temperature are different. Sintering exists throughout the whole H–O treatment and would be more serious with an increase in temperature. Moreover, the H2O generated during the H–O treatment can accelerate the sintering of Pt NPs (Fig. S4†). However, the energy required for the redispersion of Pt is sufficient when the treatment temperature reaches 600 °C and a subsequent increase in temperature has no significant influence on the further improvement of the redispersion degree. In detail, when treated at 500 °C, both the redispersion and sintering are weak, which is reflected by statistical analysis in that the size of Pt NPs with a maximum number is 1.75 nm (larger than 1.05 and 1.65 nm when treated at 600 and 700 °C, respectively) and the proportion of Pt NPs with sizes larger than 3.2 nm is just 14.7% (that would be as much as 21.5% when treated at 700 °C). During H–O treatment at 600 °C, although sintering is aggravated to some extent, redispersion is improved enormously, resulting in redispersion dominating the treatment process. It is speculated that compared with treatment at 500 °C, the formation and breakup of Pt–O–Fe or Pt–O–O–Fe bonds are much easier at 600 °C under oxidative and reductive atmospheres, respectively. When the treatment temperature reaches 700 °C, sintering aggravates greatly and the proportion of Pt NPs with sizes larger than 3.2 nm reaches 21.5% as mentioned above, which is much larger than 3% according to the statistical results of samples treated at 600 °C. Moreover, the rising temperature could not further promote the degree of redispersion obviously, leading to the fact that the size of Pt NPs with maximum number is 1.65 nm (larger than 1.05 nm when treated at 600 °C). Therefore, it could be seen that at 600 °C, sintering is not so serious and almost “overcome” by redispersion during the treatment process, leading to the final result that the proportions of Pt NPs with sizes smaller than 1.5 and 3.2 nm are 81.1% and 98.2%, respectively.
Conclusions
In this work, the redispersion behaviour of Pt on the surface of Fe2O3 was observed and studied. After treatment under alternating oxidation and reduction atmospheres at 600 °C, the average size of the Pt NPs decreased from the original 3.2 nm to 1.1 nm. Besides, we investigated the influence of treatment temperature on the redispersion of Pt and found that the optimal treatment temperature for redispersion was 600 °C. Based on a variety of characterizations, it is proposed that the Pt–O–Fe bonds formed under an oxidative atmosphere and related defects or oxygen vacancies formed under a reductive atmosphere are key factors for Pt redispersion on the surface of Fe2O3. We hope that our work can be helpful for research on anti-sintering, the regeneration of deactivated catalyst and “tuning selectivity” through changing the dispersion of noble metals.15,16,18,37–39Acknowledgements
The authors thank the National Natural Science Foundation of China. This work was supported by the National Natural Science Foundation of China (21273221).Notes and references
- L. F. Liotta, Appl. Catal., B, 2010, 100, 403–412 CrossRef CAS.
- X. Wang, C. Wang, L. Cheng, S. T. Lee and Z. Liu, J. Am. Chem. Soc., 2012, 134, 7414–7422 CrossRef CAS PubMed.
- C. Chen, C. Nan, D. Wang, Q. Su, H. Duan, X. Liu, L. Zhang, D. Chu, W. Song, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2011, 50, 3725–3729 CrossRef CAS PubMed.
- V. P. Santos, S. A. C. Carabineiro, P. B. Tavares, M. F. R. Pereira, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal., B, 2010, 99, 198–205 CrossRef CAS.
- G. Li and L. Li, RSC Adv., 2015, 5, 36428–36433 RSC.
- Z. Miao, T. Wu, J. Li, T. Yi, Y. Zhang and X. Yang, RSC Adv., 2015, 5, 19823–19829 RSC.
- Z. Miao, Y. Zhang, X. Pan, T. Wu, B. Zhang, J. Li, T. Yi, Z. Zhang and X. Yang, Catal. Sci. Technol., 2015, 5, 1314–1322 CAS.
- H. Hong, L. Hu, M. Li, J. Zheng, X. Sun, X. Lu, X. Cao, J. Lu and H. Gu, Chem. - Eur. J., 2011, 17, 8726–8730 CrossRef CAS PubMed.
- A. M. Pasqualeti, P.-Y. Olu, M. Chatenet and F. H. B. Lima, ACS Catal., 2015, 5, 2778–2787 CrossRef CAS.
- Z. Zhang, Y. Jiang, M. Chi, Z. Yang, C. Wang and X. Lu, RSC Adv., 2015, 5, 94456–94461 RSC.
- F. Diehl, J. Barbier, D. Duprez, I. Guibard and G. Mabilon, Appl. Catal., A, 2015, 504, 37–43 CrossRef CAS.
- M. O. Kazakov, A. V. Lavrenov, M. S. Mikhailova, N. A. Allert, T. I. Gulyaeva, I. V. Muromtsev, V. A. Drozdov and V. K. Duplyakin, Kinet. Catal., 2010, 51, 438–443 CrossRef CAS.
- M. O. Kazakov, A. V. Lavrenov, I. G. Danilova, O. B. Belskaya and V. K. Duplyakin, Kinet. Catal., 2011, 52, 573–578 CrossRef CAS.
- M. O. Kazakov, A. V. Lavrenov, O. B. Belskaya, I. G. Danilova, A. B. Arbuzov, T. I. Gulyaeva, V. A. Drozdov and V. K. Duplyakin, Kinet. Catal., 2012, 53, 101–106 CrossRef CAS.
- K. Morgan, A. Goguet and C. Hardacre, ACS Catal., 2015, 5, 3430–3445 CrossRef CAS.
- K. Morgan, R. Burch, M. Daous, J. J. Delgado, A. Goguet, C. Hardacre, L. A. Petrov and D. W. Rooney, Catal. Sci. Technol., 2014, 4, 729 CAS.
- J. Sa, A. Goguet, S. F. Taylor, R. Tiruvalam, C. J. Kiely, M. Nachtegaal, G. J. Hutchings and C. Hardacre, Angew. Chem., Int. Ed., 2011, 50, 8912–8916 CrossRef CAS PubMed.
- K. Morgan, R. Burch, M. Daous, J. J. Delgado, A. Goguet, C. Hardacre, L. A. Petrov and D. W. Rooney, Catal. Struct. React., 2015, 1, 120–124 CrossRef.
- H. Lieske and J. Voelter, J. Phys. Chem., 1985, 89, 1841–1842 CrossRef CAS.
- R. M. J. Fiedorow and S. E. Wanke, J. Catal., 1976, 43, 34–42 CrossRef CAS.
- R. M. J. Fiedorow, B. S. Chahar and S. E. Wanke, J. Catal., 1978, 51, 193–202 CrossRef CAS.
- T. Wu, X. Pan, Y. Zhang, Z. Miao, B. Zhang, J. Li and X. Yang, J. Phys. Chem. Lett., 2014, 5, 2479–2483 CrossRef CAS PubMed.
- Y. Nagai, N. Takagi, Y. Ikeda, K. Dohmae, T. Tanabe, G. Guilera, S. Pascarelli, M. Newton, H. Shinjoh and S. i. Matsumoto, AIP Conf. Proc., 2007, 882, 594–596 CrossRef CAS.
- M. J. D’Aniello Jr, D. R. Monroe, C. J. Carr and M. H. Krueger, J. Catal., 1988, 109, 407–422 CrossRef.
- G. Wang, H. Lian, W. Zhang, D. Jiang and T. Wu, Kinet. Catal., 2002, 43, 433–442 CrossRef CAS.
- N. An, Q. Yu, G. Liu, S. Li, M. Jia and W. Zhang, J. Hazard. Mater., 2011, 186, 1392–1397 CrossRef CAS PubMed.
- Y. El Mendili, J.-F. Bardeau, N. Randrianantoandro, F. Grasset and J.-M. Greneche, J. Phys. Chem. C, 2012, 116, 23785–23792 CAS.
- X. Zhang, Y. Niu, X. Meng, Y. Li and J. Zhao, CrystEngComm, 2013, 15, 8166–8172 RSC.
- F. Jiao, J.-C. Jumas, M. Womes, A. V. Chadwick, A. Harrison and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 12905–12909 CrossRef CAS PubMed.
- L. Zhong, J. Hu, H. Liang, A. Cao, W. Song and L. Wan, Adv. Mater., 2006, 18, 2426–2431 CrossRef CAS.
- Y. Chueh, M. Lai, J. Liang, L. Chou and Z. Wang, Adv. Funct. Mater., 2006, 16, 2243–2251 CrossRef CAS.
- I. Mitov, Z. Cherkezova-Zheleva and V. Mitrov, Phys. Status Solidi A, 1997, 161, 475–482 CrossRef CAS.
- Y. Chen, G. Xiao, T. Wang, Q. Ouyang, L. Qi, Y. Ma, P. Gao, C. Zhu, M. Cao and H. Jin, J. Phys. Chem. C, 2011, 115, 13603–13608 CAS.
- J. Lu, X. Jiao, D. Chen and W. Li, J. Phys. Chem. C, 2009, 113, 4012–4017 CAS.
- G. Sun, B. Dong, M. Cao, B. Wei and C. Hu, Chem. Mater., 2011, 23, 1587–1593 CrossRef CAS.
- A. S. Ivanova, E. M. Slavinskaya, O. A. Stonkus, R. V. Gulyaev, T. S. Glazneva, A. S. Noskov and A. I. Boronin, Catal. Sci. Technol., 2015 10.1039/c5cy01588j.
- J. Okal, H. Kubicka, L. K. epiński and L. Krajczyk, Appl. Catal., A, 1997, 162, 161–169 CrossRef CAS.
- A. Monzón, T. F. Garetto and A. Borgna, Appl. Catal., A, 2003, 248, 279–289 CrossRef.
- A. K. Aboul-Gheit, A. A. Aboul-Enein, A. E. Awadallah, S. A. Ghoneim and E. A. Emam, Chin. J. Catal., 2010, 31, 1209–1216 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01803c |
| This journal is © The Royal Society of Chemistry 2016 |