
A UV-sensitive hydrogel based combinatory drug delivery chip (UV gel-Drug Chip) for cancer cocktail drug screening†
Abstract
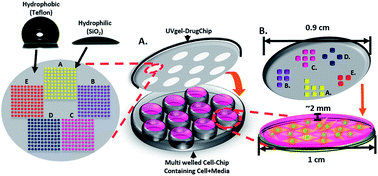
The effective and efficient treatment of diseases, such as HIV, cancer or hereditary diseases, requires accurate and precise control of the combinatorial drug-dosage and their release. Herein, we introduce a simple photosensitive poly(ethylene glycol) diacrylate (PEGDA) hydrogel based platform for high dynamic range testing of combinatorial cocktail drug screening using three chemical and two protein drug treatments for colon cancer. UV cross linked PEGDA hydrogel droplet arrays on a Teflon patterned glass substrate enable a rapid yet accurate selection and dosage assignment of the drugs. Precisely loaded cocktails of the anticancer drugs were simultaneously released in-parallel with the PEGDA hydrogel chips into 2D or 3D cultured HCT-8 colon cancer cells for combinatorial drug screening. We demonstrate the functionality of our UV gel-Drug Chips 1000 fold range of concentrations for each of the five drugs in 30 seconds to find the optimized drug cocktail using a fractional factorial control system. Our device has low drug consumption, requiring only 12 nL per screening run per droplet. In addition, our UV gel-Drug Chips were employed for find the optimized drug cocktail using a fractional search algorithm. Our cocktail drug response results for both 2D (cell viability is 7.3%) and 3D (cell viability is 10.8%) colon cancer cells were analogous to those found by conventional method (6.8 and 9.3 respectively). In contrast to conventional method, our approach is faster, more effective, less time consuming and requires a lower amounts of drug volume.
 Please wait while we load your content...
Please wait while we load your content...

