
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structure and coordination properties of novel bifunctional carboxylic derivatives of 1,3-alternate tetrathiacalix[4]arene†
Sergey N.
Podyachev
*a,
Gulnaz Sh.
Gimazetdinova
b,
Aidar T.
Gubaidullin
a,
Victor V.
Syakaev
a,
Svetlana N.
Sudakova
a,
Bulat M.
Gabidullin
a,
Vladimir T.
Ivanov
a,
Edward L.
Gogolashvili
a and
Alexander I.
Konovalov
a
aA. E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center of Russian Academy of Sciences, Arbuzov str., 8, 420088, Kazan, Russia. E-mail: spodyachev@iopc.ru; Fax: +7-843-273-1872
bKazan National Research Technological University, K. Marks str., 68, 420015, Kazan, Russia
First published on 10th February 2016
Abstract
New bifunctional derivatives of 1,3-alternate tetrathiacalix[4]arene decorated with carboxylic, ester, hydrazide and/or hydrazone groups have been synthesized with good yields using the tetrathiacalix[4]arene derivatives with incorporated pairs of carboxylic and ester groups as versatile building blocks. The structural peculiarities of the obtained bifunctional compounds have been investigated by means of X-ray analysis, IR and NMR spectroscopy. The recognition ability of the synthesized macrocycles towards some alkali, alkali-earth and transition metal ions has been investigated applying a solvent extraction method. The results showed that the structure of a calix[4]arene platform as well as the nature of functional substitutes located on opposite sides of the macrocycle are critical for the coordination properties of the synthesized compounds.
1. Introduction
The design and synthesis of artificial receptors for cation and anion guests is an exciting topic of current chemistry because of the extreme importance of these compounds for coordination and supramolecular chemistry as well as for biological processes involving molecular recognition of cationic, anionic and neutral species.1–7 The sensing properties of the receptors are usually based on their ability to form multiple-point binding sites during interaction with a variety of substrates. It is well-known that macrocyclic compounds with several donor centers can reveal a unique selectivity towards some substrates. However, there are some restrictive factors on the way of preparing such type of receptors and they are connected not only with an accurate simulation of the structure or prediction the recognition properties of these compounds but obviously deal with the problem of their further synthesis. In many cases, the solving of this problem opens new possibilities to the successful construction of artificial receptors. At the same time, current industrial needs require more and more advanced compounds possessing unique properties and enabling the progress in the creation of novel functional materials. Therefore, a search for rational approaches to the synthesis of ligands with a desired set of properties is an actual task.One of the strategies for the molecular design of ligands can be founded on the molecular LEGO approach. It assumes that even a trivial combination of the basic molecular platforms and functional building blocks makes possible to obtain a great number of various sophisticated structures with desired and predictable properties, applying, as a rule, relatively simple chemical protocols. The calix[n]arenes are known as a synthetically and commercially accessible class of compounds which can be successfully applied for these purposes due to their suitable molecular scaffolds.8,9 A distinctive feature of this class of macrocyclic compounds is their three-dimensional spatially preorganized structure that can be fixed in various isomeric forms. Moreover, such compounds give an opportunity for incorporating of a variety of substitutes both at the upper and lower rims. These peculiarities of calix[4]arenes provide their application as keystones for the creation of bifunctional receptors having unequal binding centers.10–12 A possibility of coordinating at least two types of substrates simultaneously extends an application area of these compounds. An interest to such type of calix[4]arene derivatives can be also explained by the fact that they can reveal the biomimetic properties.13 Additionally, a presence of spatially divided binding and reaction sites in the structure of the compounds gives an excellent opportunity for the preparation on their base the polymeric as well as covalently fixed on a hard substrate sensing materials.
Among calix[n]arene family compounds, the tetrathiacalix[4]arenes attract a special attention. In opposite to their classical analogues, they can be easily fixed in any of cone, partial cone or 1,3-alternate isomer forms by using a template effect of metal cations just on the initial stage of their functionalizing. Although the cone-isomers usually reveal substantially higher binding efficiency towards different ions in comparison with 1,3-alternate isomers, the latter are interesting due to their enhanced binding selectivity.14,15 Furthermore, the 1,3-alternate tetrathiacalix[4]arenes can be considered as very suitable tectons for the construction of artificial heteroditopic receptors capable of exhibiting a mimic allosteric binding that plays a major role in biological systems.11,12,16,17
In present work we demonstrate a synthetic strategy for the preparing of novel 1,3-alternate tetrathiacalix[4]arene derivatives functionalized by pairs of carboxylic, ester, hydrazide, chloroanhydride as well as hydrazone groups and report the details of the synthesis and spectroscopic characterizations of these compounds. Their structural peculiarities have been investigated by means of X-ray analysis. The receptor properties of the synthesized bifunctional derivatives against alkali, alkali-earth and transition metals were studied by liquid–liquid extraction method and are discussed below.
2. Results and discussion
2.1. Synthesis
The compounds with ester and carboxylic groups often serve as starting compounds for a further synthesis of a wide range of organic compounds. Therefore, the calix[4]arene scaffolds functionalized by both these groups can be potentially used as promising building blocks for the obtaining of bifunctional macrocyclic derivatives due to the different reactivity of ester and carboxylic groups.18,19 The synthetic route utilized by us for the preparation of bifunctional compounds is presented in Fig. 1. The synthesis starts from the parent tetrathiacalix[4]arenes 1a and 1b which are converted into the corresponding tetraethyl esters 2a and 2b after their alkylation with ethyl bromoacetate in the refluxing acetone and using Cs2CO3 as a base.20,21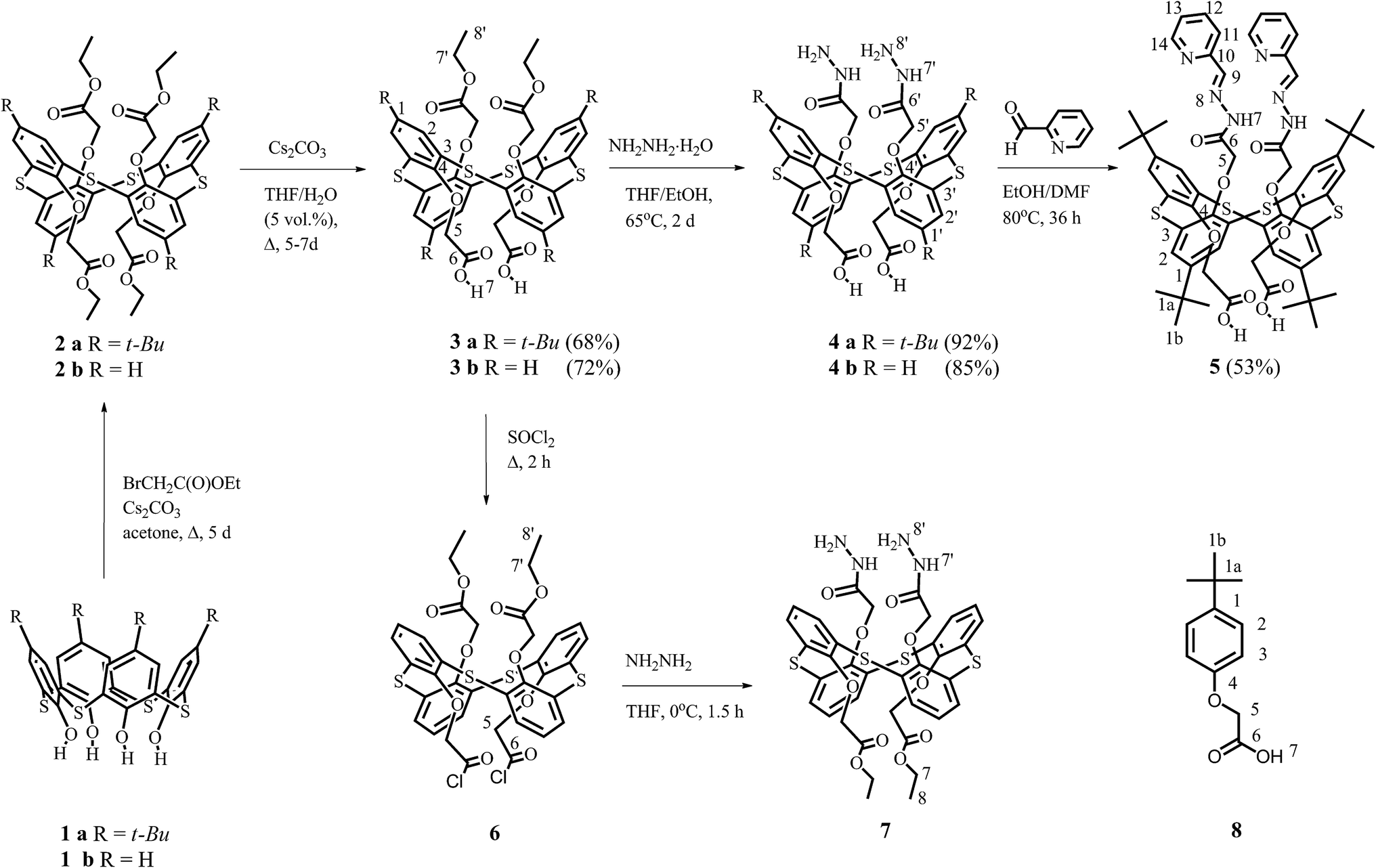 | ||
| Fig. 1 Synthetic routes and structural formulae of the investigated compounds. Numbering system of atoms used in the experimental section. | ||
1,3-Alternate tetrathiacalix[4]arenes are known to bind Cs+ ions only by “one side” of the macrocycle. This selectivity is obviously caused by the allosteric effect.22,23 Taking this fact into account, we have developed an effective protocol for the synthesis of dicarboxylic tetrathiacalix[4]arene derivatives 3a and 3b. The reflux of the compounds 2a and 2b in the THF solution containing 5 vol% of water and 12 equivalents of Cs2CO3 resulted in the formation of cesium salts of 3a and 3b, respectively. After treatment of the precipitates with HCl, the calix[4]arenes 3a and 3b were obtained with good yields. The details of the synthetic procedure and the supposed mechanism of the reaction as well as the structures of the compounds 3a, 3b and dicesium salt of 3a determined by the X-ray analysis have been reported recently.24
The obtained compounds 3a and 3b can undergo a further selective transformation on both sides of the tetrathiacalix[4]arene platform. We were especially interested in the introduction of hydrazide groups into the macrocyclic framework. According to the investigations accomplished in the last decade, the hydrazide derivatives of calix[4]arene can act as selective receptors for anions and cations.14,15,25–27 Furthermore, these compounds can be successfully applied as key reagents for the synthesis of various nitrogen containing derivatives, including acylhydrazones, acylsemicarbazones and heterocycles.
The tetrathiacalix[4]arenes 4a and 4b functionalized by acetylhydrazide groups have been prepared in 92% and 85% yields by the reflux of the corresponding tetrathiacalixarenyloxyacetic acid diesters 3a and 3b with an excess of NH2–NH2·H2O in the THF-EtOH solution for 2 days (Fig. 1). The crude products isolated from the reaction mixture can include the hydrazine molecules captured due to the interactions with carboxylic groups of the tetrthiacalix[4]arene molecules. Therefore, for obtaining the hydrazine free products 4a and 4b, the solid remainders were suspended in water and thereafter treated carefully with HCl to reach pH ∼5.
The condensation of dihydrazide 4a with picolinaldehyde has been accomplished in DMF–EtOH solution by heating the mixture at 80 °C over 36 h. After an appropriate treatment, the target dihydrazone 5 was obtained with the yield of 53%.
The synthesis of diester–dihydrazide derivative 7 started from the diester–dicarbonic acid 3b. At the first stage, 3b was converted into dichloroanhydride derivative 6 under treatment with thionyl chloride and reflux the mixture for 2 h. A subsequent condensation of the crude product 6 with an anhydrous hydrazine dispersed in the absolute THF at 0 °C led to the formation of bifunctional derivative 7 with the yield 72%. The synthesis should be carried out with a special accuracy, since a variety of by-products can be formed during the reaction. It should be also noted that we have attempted to obtain the compound 7 by the etherification of 4b in the presence of EtOH and p-toluenesulfonic acid, as a catalyst. However, the yield of a target product did not exceed 30%.
In order to explore a complexation behavior of novel synthesized compounds, the monomeric carboxylic counterpart 8, as a structural block of tetrathiacalix[4]arenes 3–5, has been obtained from p-tert-butyl-phenoxyacetic acid ester.28
2.2. IR characterization in solid
IR spectra of p-tert-butyltetrathiacalix[4]arenes 3a, 4a, 5 differ from the spectra of tetrathiacalix[4]arenes 3b, 4b, 6 and 7 due to the presence in former of the intensive bands ν(CH3) at ∼2960 cm−1 assigned to the tert-butyl groups enclosed in the structure of the compounds (see Table 1 and Fig. 1, 2, 5, 8, 11, 14 and 17 in ESI†). In IR spectra of the compounds 3a and 3b, the absorption bands νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O belonging to the ester groups have almost the same maxima (1767 cm−1 and 1762 cm−1). At the same time, the absorption bands νCO of carboxylic groups for 3a appear as two peaks at 1738 cm−1 and 1692 cm−1. In the case of 3b, we observe a single but rather intensive peak at 1723 cm−1 in the spectrum obtained in KBr pellets. The second peak having a weak intensity and low-frequency was detected at 1663 cm−1 only in Nujol. The presence of both peaks in the spectra of 3a and 3b indicates obviously an occurrence as free carboxylic groups in the structure of the compounds and the groups participating in the hydrogen bonding as well.
O belonging to the ester groups have almost the same maxima (1767 cm−1 and 1762 cm−1). At the same time, the absorption bands νCO of carboxylic groups for 3a appear as two peaks at 1738 cm−1 and 1692 cm−1. In the case of 3b, we observe a single but rather intensive peak at 1723 cm−1 in the spectrum obtained in KBr pellets. The second peak having a weak intensity and low-frequency was detected at 1663 cm−1 only in Nujol. The presence of both peaks in the spectra of 3a and 3b indicates obviously an occurrence as free carboxylic groups in the structure of the compounds and the groups participating in the hydrogen bonding as well.
| Compound | Vibration assignment | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ν asNH2NH | ν sNH2NH | δNH | δNH2 |
νCO |
νPh | νCH2(3) | νCSC | ||
| a Additional characteristics of absorption bands: sh – shoulder; br – broad; vbr – very broad. The maxima of rather intensive bands of signals are solid underlined. The maxima of weak intensive bands of signals are dashed underlined. b Characteristic vibrations are not due to overlap with Nujol bands. c Absorption band no detected. d Characteritic vibrations of νNH. | |||||||||
| 3a | KBr | 1767 | 1450 | 2959 | 1050 br | ||||
| 1738 | 2907 | 1090 | |||||||
| 1692 | 2872 | ||||||||
| Nujol | 1766 | 1460 br | —b | 1053 | |||||
| 1738 | 1068 | ||||||||
| 1690 br | 1089 | ||||||||
| 3b | KBr | 1762 | 1419 vbr | 2981 | 1054 | ||||
| 1723 br | 2903 | 1078 | |||||||
| Nujol | 1761 | 1462 br | —b | 1058 | |||||
| 1724 br | 1437 sh | 1078 | |||||||
| 1663 br | 1417 sh | ||||||||
| 4a | KBr | 3423 br | 1541 br | 1626 sh | 1758 | 1478 sh | 2963 | 1041 | |
| 3333 | 1682 br | 1450 | 2909 | ||||||
| 1430 | 2872 | ||||||||
| Nujol | 3422 br | —c | 1627 | 1758 | 1457 br | —b | 1041 br | ||
| 3330 br | 1681 vbr | ||||||||
| 4b | KBr | 3416 br | 3210 sh | 1522 br | 1620 sh | 1718 sh | 1418 | 2912 | 1039 br |
| 3319 br | 1683 vbr | ||||||||
| Nujol | 3415 br | 3210 br | 1522 | 1620 sh | 1720 sh | 1461 | —b | 1041 br | |
| 3318 | 1685 vbr | 1436 | |||||||
| 1418 | |||||||||
| 5 | KBr | 3445 vbrd | 1534 | 1757 sh | 1467 | 2962 | 1044 br | ||
| 3329d | 1707 br | 1448 | 2907 | ||||||
| 3234d | 1429 | 2872 | |||||||
| Nujol | 3459 brd | 1537 | 1758 | 1463 br | —b | 1046 | |||
| 3293d | 1717 vbr | ||||||||
| 6 | KBr | 1815 | 1474 | 2989 | 1060 | ||||
| 1802 | 1422 | 2903 | |||||||
| 1766 | 1408 | ||||||||
| Nujol | 1815 | 1462 | —b | 1060 | |||||
| 1802 | 1422 | ||||||||
| 1766 | 1408 | ||||||||
| 7 | KBr | 3429 | 3210 sh | 1505 | 1632 | 1765 | 1473 | 2981 | 1038 br |
| 3328 vbr | 1729 | 1420 br | 2906 | ||||||
| 1677 br | |||||||||
| Nujol | 3428 | 3210 | 1502 | 1632 sh | 1728 | 1462 br | —b | 1039 | |
| 3336 br | 1675 br | 1422 sh | |||||||
The replacement of ester groups in the compounds on the hydrazide (for 4a and 4b) or the hydrazone (for 5) ones leads to the appearance (>3000 cm−1) of complicated absorption bands in their spectra caused by νNH vibrations. The similar peaks are observed in the spectrum of dihydrazide derivative 7 as well. The presence of amide-II absorption band (δNH ∼ 1520–1540 cm−1) in the spectra of 4a, 4b, 5 and 7 indicates the trans-conformation of amide groups in the structure of these compounds.29–31 At the same time, an absorption band νNH ∼ 3234 cm−1 which is also observed in the spectrum of the dihydrazone 5 proves a contribution of cis-form to the conformational composition of the compound.
The presence of low-frequency absorption bands νasNHNH2 along side with high-frequency peaks (3423 cm−1 and 3333 cm−1 for 4a, 3416 cm−1 and 3318 cm−1 for 4b in KBr) in the spectra of the compounds points to the fact that at least one of the hydrazide groups in these molecules participates in the hydrogen bonding. A similar picture is observed in the case of dihydrazone derivative 5 (3445 cm−1 and 3329 cm−1) as well.
An occurrence of hydrazide and carboxylic groups in the structure of the compounds 4a and 4b leads to the appearance in their spectra of a broad absorption peak (at 1680 cm−1) arising due to the overlap of νCO bands. In the case of 4a, a low-intensity peak at 1758 cm−1 caused probably by the presence of free carboxylic groups is also detected. When going to the dihydrazone 5, a broad absorption band νCO with a maximum at 1707 cm−1 was observed in the spectrum of this compound as well. The underfrequency for this vibration is obviously connected with the participation of carboxylic and hydrazone groups in the hydrogen bonding. However, the hydrogen bonds, in this case, are not so strong, as those formed in the compounds 4a and 4b. In the case of spectra 6, the doublet ν(CO) at 1815 cm−1 and 1802 cm−1 for chloroanhydride groups and a sharp single peak for ester groups at 1766 cm−1 are observed in KBr. An absorbance ν(CO) of ester groups for the compound 7 is detected exactly at the same frequency (1765 cm−1) but appears as a quiet low intensive peak. At the same time, an absorbance of νCO of hydrazide groups is revealed as two peaks having an equal intensity at 1729 cm−1 and 1677 cm−1, indicating the involving only one of the carbonyl oxygen atoms of these groups in the hydrogen bonding.
The hydroxyl groups appear as a broad dome-shaped ν(OH) band in IR spectra of the compounds 3a, 3b, 4a and 4b. This band is underneath the ν(CH), ν(–CH) and ν(NH) absorptions in the region ∼3500–3100 cm−1 with a center being around ∼3400 cm−1 for 3a, 3b and ∼3250 cm−1 for 4a and 4b. These values of frequency are lower than ones observed for the stretching vibrations of free hydroxyl groups (∼3500 cm−1), that proves their participation in the formation of H-bonds. In the case of dihydrazone derivative 5, the absorbance ν(OH) has more broadened shape with an uncertain maximum.
2.3. X-ray analysis
Suitable for X-ray analysis the colorless crystals of dihydrazides 4a and 4b were obtained by the recrystallization of their crude products from MeOH–CH2Cl2. We have also succeeded in the preparing the crystals of 4a in DMSO when treated a crude product with HCl (see Experimental part). In former case, 4a and 4b calix[4]arenes were crystallized with the capturing two and one hydrazine molecules, affording consequently the structures 4a·2NH2NH2 (Fig. 2a) and 4b·NH2NH2 (Fig. 2c). In the second case, the crystals of 4a were obtained without inclusion the hydrazine in their structure (Fig. 2b). The recrystallization of dihydrazide 7 from MeOH–CH2Cl2 has also brought the crystals do not containing hydrazine molecules (Fig. 2d). In the last case, the product was treated by water without adding HCl. The presence of carboxylic groups in 4a and 4b in contrast to 7 is obviously a main reason for the hydrazine binding by these compounds. The acidification of 4a and 4b solutions prevents the amine coordination. The crystals of dihydrazone 5 were obtained from MeOH–CH2Cl2 as well. All considered bifunctional derivatives 4a, 4b, 5 and 7 possess the 1,3-alternate isomer form established for their parent 3a and 3b tetrathiacalix[4]arenes previously. | ||
| Fig. 2 The structures of compounds (a) 4a·2NH2NH2; (b) 4a; (c) 4b·NH2NH2 and (d) 7 in the crystals. Only hydrogen atoms of OH and NH groups are shown. The disordered fragments are shown in positions with the highest occupancy. In the cases of (a) – the hydrazine molecule, (b) – the DMSO molecule and (c) – the water molecules outside of the thiacalix[4]arene cavity are not shown. | ||
The compound 4a·2NH2NH2 crystallizes in C2/c space group with one hydrazine molecule being in thiacalix[4]arine's cavity and the another one being outside (Fig. 2a). The highly disordered solvent molecules can be localized between thiacalix[4]arene molecules in the crystal and occupy up to 1307 Å3 per unit cell. However, it should be noticed that these solvate molecules could not be detected properly using Fourier electron density synthesis under normal X-ray experiment conditions. A coordination of hydrazine molecules by the thiacalix[4]arene 4a leads to an increase only one from four of dihedral angles formed by the aromatic moieties and a reference plane passing through sulfur atoms (for compare in 4a: 119.1(2)°, 113.1(2)°, 116.0(2)° and 116.0(2)°; in 4a·2NH2NH2: 127.4(2)°, 109.1(2)°, 105.7(2)° and 108.3(2)°).
The hydrazine free thiacalix[4]arene 4a crystallizes in P21/n space group with one DMSO molecule per macrocycle directed outside of the cavity (Fig. 2b). All tert-butyl groups are disordered over two positions with relative occupancies: 0.77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.23, 0.60:0.40, 0.65:0.35 and 0.65:0.35. The carboxylic group is also disordered over two positions with a relative occupancy being 0.57:0.43. Inequality of bond lengths in carboxylic groups indicate that both –COOH substituents in the molecule are not deprotonated.
:0.23, 0.60:0.40, 0.65:0.35 and 0.65:0.35. The carboxylic group is also disordered over two positions with a relative occupancy being 0.57:0.43. Inequality of bond lengths in carboxylic groups indicate that both –COOH substituents in the molecule are not deprotonated.
The realization of classical hydrogen bonds in the molecules of thiacalix[4]arene 4a results in the formation of H-dimers in the crystals due to the interaction between hydrazide groups of the centro-symmetrical pairs of molecules, whereas the carboxylic groups do not form intra- and intermolecular hydrogen bonds between each other. It is obviously caused by the fact that one of the carboxylic groups is involved in the hydrogen bonding with DMSO molecule located in the macrocycle cavity, and the another one participates in the intramolecular hydrogen bonding with ester oxygen of an opposite phenoxylic fragment of the thiacalix[4]arene framework.
The compound 4b·NH2NH2 crystallizes in P21/c space group. The asymmetric part of the unit cell contains one thiacalix[4]arene molecule together with one hydrazine molecule being inside (Fig. 2c), as in the case of 4a·2NH2NH2, and four co-crystallized water molecules located outside the cavity. Dihedral angles between aromatic moieties and a reference plane passing through sulfur atoms (109.3(1)°, 104.1(1)°, 100.7(1)° and 123.6(1)°) are slightly less than the corresponding values for 4a. An absence of bulky tert-butyl groups at the upper rim of thiacalix[4]arene favours the tube-shaped structure of 4b which is clear observed for this compound even under encapsulation.
Insufficiently high quality of 4a·2NH2NH2 and 4b·NH2NH2 crystals prevents a deep analysis of the bond distances and angles in these molecules as well as makes difficult a precise determination of the hydrogen atoms in OH and NH2 groups. Nevertheless, it is clearly seen that the hydrazine molecule is coordinated due to the participation of both carboxylic groups. It should be also noticed that dicarboxylic tetrathiacalix[4]arene derivatives can generate a formation of salt structures with an involvement of deprotonated carboxylic group of macrocycle and the hydrazinium ion.
The dihydrazide 7 crystallizes in P21/c space group. The asymmetric part of the unit cell comprises just one thiacalix[4]arene molecule and does not enclose solvate molecules (Fig. 2d). The dihedral angles between the aromatic moieties and the plane defined by four sulfur atoms were determined as 111.5(1)°, 99.1(1)°, 105.2(1)° and 115.8(1)°. One of the hydrazide and one of the ester groups are disordered over two positions with relative occupancy ratios of 0.45:0.55 and 0.42:0.58, respectively. The hydrazide substitutes are involved in intramolecular as well as the intermolecular hydrogen bonds simultaneously, which results in 3D supramolecular structure formed by 7 in the crystal.
The dihydrazide derivatives 4a, 4b and 7 have trans-amide conformation of the acetylhydrazide fragments and the non-hydrogen atoms of these groups are located practically at the same plain. One of the hydrazide substituents closes a pseudo-cavity of the thiacalix[4]arene and prevents a penetration of solvent molecules (Fig. 2a–d). Moreover, this spatial structure of the molecules is additionally stabilized by a system of intramolecular and intermolecular hydrogen bonds.
Dihydrazone derivative 5 crystallizes in P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The asymmetric part of the unit cell contains one thiacalix[4]arene molecule and one methanol molecule which acts as a bridge between two hydrazone fragments (Fig. 3). Dihedral angles between aromatic moieties and a reference plane passing through sulfur atoms are 116.9(5)°, 103.7(5)°, 105.3(5)° and 110.3(4)°. One of the tert-butyl groups is disordered over two positions with a relative occupancy ratio of 0.54:0.46.
space group. The asymmetric part of the unit cell contains one thiacalix[4]arene molecule and one methanol molecule which acts as a bridge between two hydrazone fragments (Fig. 3). Dihedral angles between aromatic moieties and a reference plane passing through sulfur atoms are 116.9(5)°, 103.7(5)°, 105.3(5)° and 110.3(4)°. One of the tert-butyl groups is disordered over two positions with a relative occupancy ratio of 0.54:0.46.
 | ||
| Fig. 3 The geometry of the molecule (a) and H-dimer (b) in the crystal of 5 in the crystal. Hydrogen atoms are not shown, except for the hydrazone and carboxyl groups of 5 and MeOH. | ||
In the case of tetrathiacalix[4]arene 5, both acetylhydrazone substitutes are characterized by the flattened structure and are described by EN–NECN configuration which is typical for acetylhydrazones in crystal.32 At the same time, one of the acetylhydrazone fragments has cis-conformation and the another one is in the trans-amide form. The previously investigated tetra-acetylhydrazone derivatives of thiacalix[4]arene were also characterized by an equal ratio of cis/trans-amide conformers. However, the acetylhydrazones, according to CSD data, prefer the trans-amide conformation (94% of all structures).32 A mutual influence of the hydrazone fragments immobilized on the macrocyclic platform leads obviously to an equalization of the conformational content. The acetylhydrazone fragments are interconnected directly via the intramolecular hydrogen bonds and by means of the bridge interaction with participation of the hydroxyl groups of MeOH as well.
In spite of the fact that strong donor and acceptor groups are present in the molecule 5, only pair interactions between the hydroxyl groups of carboxylic substitutes of one molecule and the pyridine nitrogen atoms of a neighboring molecule are realized in the crystal. These interactions result in the formation of centrosymmetric H-dimers (or pseudo-tetramers, if two solvate MeOH molecules are taking into account). Such type of the molecules' arrangement, however, does not lead to the closest packing. Indeed, the voids revealed in the crystals occupy up to 246 Å3 per unit cell and can be filled up by the highly disordered solvent molecules.
2.4. NMR characterization in solution
The 1H and 13C spectra of bifunctional derivatives 4a, 4b, 6 and 7 as well as the spectra of starting compounds 3a and 3b described by us earlier24 show two sets of signals assigned to tetrathiacalix[4]arene scaffold (Table 2 and 3), although each of –OCH2R pairs of functional groups produces only one combination of signals. Such spectral picture for the synthesized compounds is in a full agreement with their expected structure and is typical for the distal location of the identical substitutes in molecule.| a Numbering according to Fig. 1. b Assignment for structural fragment of tetrathiacalix[4]arene containing the indicated functional group. c Registrated only trans-form. d Assignment for trans–(cis)-form of hydrazine fragments. e No detected signal. | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | 4a | 4b | 5 | 6 | 7 | 8 | |||||
| Solvent | DMSO-d6 | DMSO-d6 | DMSO-d6 | CDCl3 | DMSO-d6 | DMSO-d6 | |||||
| Fragmentb | –COOH | –C(O)NHNH2c | –COOH | –C(O)NHNH2c | –COOH | –NHNC–Py |
–C(O)Cl | –COOEt | –COOEt | –C(O)NHNH2c | –COOH |
| H1b | 1.212 | 1.200 | 1.122 | 1.244, (1.033; 1.104)d | 1.249 | ||||||
| 1.152 | |||||||||||
| 1.216 | |||||||||||
| 1.229 | |||||||||||
| H1 | 6.990, t, 3J = 7.7 | 6.964 | 6.81, t, 3J = 7.5 | 6.82, t,3J = 7.4 | 7.026, t, 3J = 7.7 | 6.984, t, 3J = 7.8 | |||||
| H2 | 7.389 | 7.386 | 7.461, d, 3J = 7.7 | 7.488 | 7.382 | 7.456 | 7.50, d, 3J = 7.5 | 7.38, d, 3J = 7.4 | 7.453, d, 3J = 7.7 | 7.447 d, 3J = 7.8 | 7.285, d, 3J = 8.76 |
| 7.481 | 7.481 | ||||||||||
| 7.498 | (7.616; 7.625)d | ||||||||||
| H3 | 6.816, d, 3J = 8.76 | ||||||||||
| H5 | 4.316 | 4.283 | 4.429 | 4.446 | 4.356 | 3.870; 4.318 (5.038; 5.203)d | 4.95 | 4.58 | 4.487 | 4.437 | 4.616 |
| 4.385 | |||||||||||
| 4.428 | |||||||||||
| 4.452 | |||||||||||
| 4.505 | |||||||||||
| H7 | —e | 7.59 | —e | 7.9c | 12.37 | 9.865; 11.104 (11.62; 11.799)d | 4.23, q,3J = 6.9 | 4.088, q, 3J = 7.1 | 7.231 | 12.917 | |
| H8 | 1.28, t, 3J = 6.9 | 1.164, t, 3J = 7.1 | —e | ||||||||
| H9 | 8.087; 8.459, (7.959; 7.978)d | ||||||||||
| H11 | 7.908 (7.716)d | ||||||||||
| H12 | 7.786 (7.860)d | ||||||||||
| H13 | 7.346 (7.432)d | ||||||||||
| H14 | 8.625 (8.541)d | ||||||||||
| a Numbering according to Fig. 1. b Assignment for structural fragment of tetrathiacalix[4]arene containing the indicated functional group. c Registrated only trans-form. d Assignment for trans–(cis)-form of hydrazine fragment. | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | 4a | 4b | 5 | 6 | 7 | 8 | |||||
| Solvent | DMSO-d6 | DMSO-d6 | DMSO-d6 | CDCl3 | DMSO-d6 | DMSO-d6 | |||||
| Fragmentb | –COOH | –C(O)NHNH2c | –COOH | –C(O)NHNH2c | –COOH | –NHNC–Py |
–C(O)Cl | –COOEt | –COOEt | –C(O)NHNH2c | –COOH |
| C1a | 31.0 | 30.7 | 33.88 | 33.7 (34.1)d | 33.7 | ||||||
| C1b | 34.1 | 33.9 | 30.70 | 30.3 (30.6)d | 31.3 | ||||||
| C1 | 147.0 | 147.2 | 123.9 | 124.8 | 146.2 | 146.6 (146.7)d | 124.3 | 124.4 | 124.1 | 124.9 | 143.1 |
| C2 | 128.8 | 129.8 | 134.8 | 134.1 | 130.5, 131.7 | 132.5 (134.0)d | 137.6 | 137.2 | 134.4 | 133.9 | 125.9 |
| C3 | 127.1 | 126.9 | 127.7 | 128.2 | 127.0 | 127.4 (127.3)d | 128.3 | 128.5 | 127.8 | 128.0 | 113.8 |
| C4 | 154.5 | 155.3 | 158.8 | 157.6 | 155.3, 155.8 | 157.4 (156.7)d | 158.3 | 159.4 | 158.4 | 157.6 | 155.4 |
| C5 | 65.6 | 68.3 | 67.6 | 67.5 | 67.3, 68.3, 68.6 | 69.6; 68.7, (67.3)d | 74.6 | 68.8 | 67.6 | 67.9 | 64.4 |
| C6 | 168.6 | 165.5 | 168.9 | 165.7 | 168.8, 169.0 | 163.1; 164.0, (168.6; 168.9)d | 168.4 | 168.3 | 167.5 | 165.4 | 170.2 |
| C7 | 60.9 | 60.4 | |||||||||
| C8 | 14.3 | 13.8 | |||||||||
| C9 | 148.4 (143.2)d | ||||||||||
| C10 | 152.8 (153.1)d | ||||||||||
| C11 | 120.2 (119.9)d | ||||||||||
| C12 | 136.7 (136.8)d | ||||||||||
| C13 | 124.4 (124.0)d | ||||||||||
| C14 | 149.4 (149.3)d | ||||||||||
The determination of the isomer form for the “classical” calix[4]arenes containing methylene bridges in their structure can be easily performed by using a simple “de Mendoza rule”.8,33 In the case of tetrathiacalix[4]arenes, a more sophisticated analysis of the chemical shifts for other groups of atoms in comparison with the model compounds as well as 2D NMR experiments are usually required. However, in our case, the situation is simplified due to the fact that n-butyl groups are just bulky enough to prevent rotation of the alkylated phenolic rings through the main annulus of thiacalix[4]arene, at least at temperatures up to 413 K.34 Since the synthetic pathway to the desired compounds started from the conformationally immobilized 1,3-alternate precursors 3a and 3b with well documented structures,24 we have assumed that their derivatives 4a, 4b, 6 and 7 would also adopt the 1,3-alternate isomer form. The 2D NOESY experiments for the compounds 4–7 have been nevertheless accomplished to confirm this fact. Really, the cross-peaks between aromatic and tert-butyl protons and the protons of OCH2– groups belonging to the acid or hydrazide fragments of the adjacent structural blocks of tetrathiacalix[4]arene were detected in the spectrum of 4a. It is quite clear that only in the case of the 1,3-alternate isomer, these cross-peaks could be observed. The similar picture was characteristic for all synthesized compounds 4–7.
It could be expected that an occurrence of amide fragments in the structures of 4a, 4b, 5 and 7 should result in the complication of their spectra due to the realization of cis- and trans-amide conformers for these compounds. In the case of compound 5, a formation of additional spatial forms is also possible due to E/Z isomerization relative to the CN double bond. Indeed, a great number of peaks as well as the broadening and overlapping signals are observed in 1H NMR spectrum of dihydrazone 5 at 303 K (Fig. 4). The various spatial forms for hydrazones have been analyzed carefully and discussed in detail in our previous publications.35,36 In accordance with the spectral criteria suggested in these articles, we have performed a conformational assignment for the investigated compounds and represented the results in the Tables 2 and 3
 | ||
| Fig. 4 The influence of the temperature change 303 K → 403 K on 1H NMR spectra of the dihydrazone tert-butyltetrathiacalix[4]arene 5. | ||
It was established that only trans-amide conformer is realized for dihydrazides 4a, 4b and 7. The hydrazone fragments in 5 were found to exist in ECN isomer form. It is worth noting that the trans-form content for the compound 5 equals 42% and is practically similar to the conformational composition determined for the corresponding 4-tert-butylphenoxyacetylhydrazone (40%)35 as well as for 1,3-alternate tetrahydrazone p-tert-butyltetrathiacalix[4]arene (44%).32
It was shown in our previous investigations that UV irradiation of 4-tert-butylphenol and resorcinol acetylhydrazone derivatives resulted in a partly conversion of the ECN into the ZCN isomer.31,35 According to NMR data, the stabilization of ZCN isomer in these compounds was provided due to the occurrence of intramolecular hydrogen bonds between NH protons and the nitrogen atoms of 2-pyridinyl substitutes in the hydrazone fragments. However, the irradiation of CDCl3 solution of the compound 5 by UV light for 2 h did not lead to the ECN/ZCN isomerization. Such situation was observed by us earlier for the calix[4]resorcinol and tetrathiacalix[4]arene (cone and 1,3-alternate) acetylhydrazone derivatives also.31,36 The presence of neighboring acetylhydrazone groups capable of participating in intramolecular interactions as well as the sterical hindrances realized in tetrathiacalix[4]arene and calix[4]resorcine macrocycles prevent evidently the UV-induced ECN/ZCN isomerization.
An occurrence of a large number of polar groups possessing acceptor and donor properties makes possible for the investigated compounds a formation of dimer and even substantially greater molecular aggregates in low-polarity solvents. It could be expected that the compounds 4a and 4b bearing the carboxylic and hydrazide groups capable of formation of a various type of intermolecular hydrogen bonds are greatly predisposed to the generation of such aggregates. To check this assumption, we have applied a diffusion NMR method which is commonly used for studying an aggregation in host–guest systems37 and investigating of the hydrogen-bonded assemblies in the solution-phase.38,39
NMR DOESY experiments performed for the compounds 4a and 4b in the mixture of CDCl3–CD3OD (1:1) have demonstrated that the investigated bifunctional derivatives does not form even dimers at the concentrations up to 30 mM. The analysis of a change of self-diffusion coefficients under increase of the ligand's concentration showed that the amount of the dimeric molecules for these compounds does not exceed 5%. It should be noticed that we have previously investigated the aggregation properties of dicarbonic acid 3b possessing a dimeric structure in the crystal.24 It appeared to our surprise that in CDCl3 solution the percentage of 3b molecules adopting the dimer form was not above 25%.
To summarize NMR experiments, we can conclude that the complication of NMR spectra for the compounds 4–7 may be caused, first of all, by the intramolecular interactions of functional groups and, in the case of the compound 5, by cis/trans amide conformational isomerism. The magnetic anisotropy of aromatic rings in 1,3-alternate isomer may additionally complicate the spectra of these compounds. This influence is especially noticeable for the compound 5. Thus, the signals of methylene groups (H5) of acetylhydrazone fragments having trans-conformation differ on 0.45 ppm (Table 2 and Fig. 4). Under heating the samples up to 373 K, the proton peaks of carboxylic groups as well as the peaks of hydrazone group protons for different conformers coalesce and converge into sharp lines at 403 K. Under cyclic temperature changes 303 K→ 403 K → 303 K, the spectral picture does not change, which testifies to the maintenance of the structural content for the compound 5.
2.5. Extraction studies
The liquid–liquid extraction experiments were performed to examine the efficiency and selectivity of tetrathiacalix[4]arene derivatives 3a, 3b, 4a, 4b, 5, 7 and their monomeric counterpart 8 in transferring of s- (Na+, Ca2+), p- (Pb2+) and d- (Co2+, Ni2+, Cu2+, Zn2+, Ag+ and Cd2+) metal ions from aqueous phase into chloroform at various pH (Fig. 5). The concentrations of ligands in chloroform and metal cations in aqueous phase were identical in all experiments.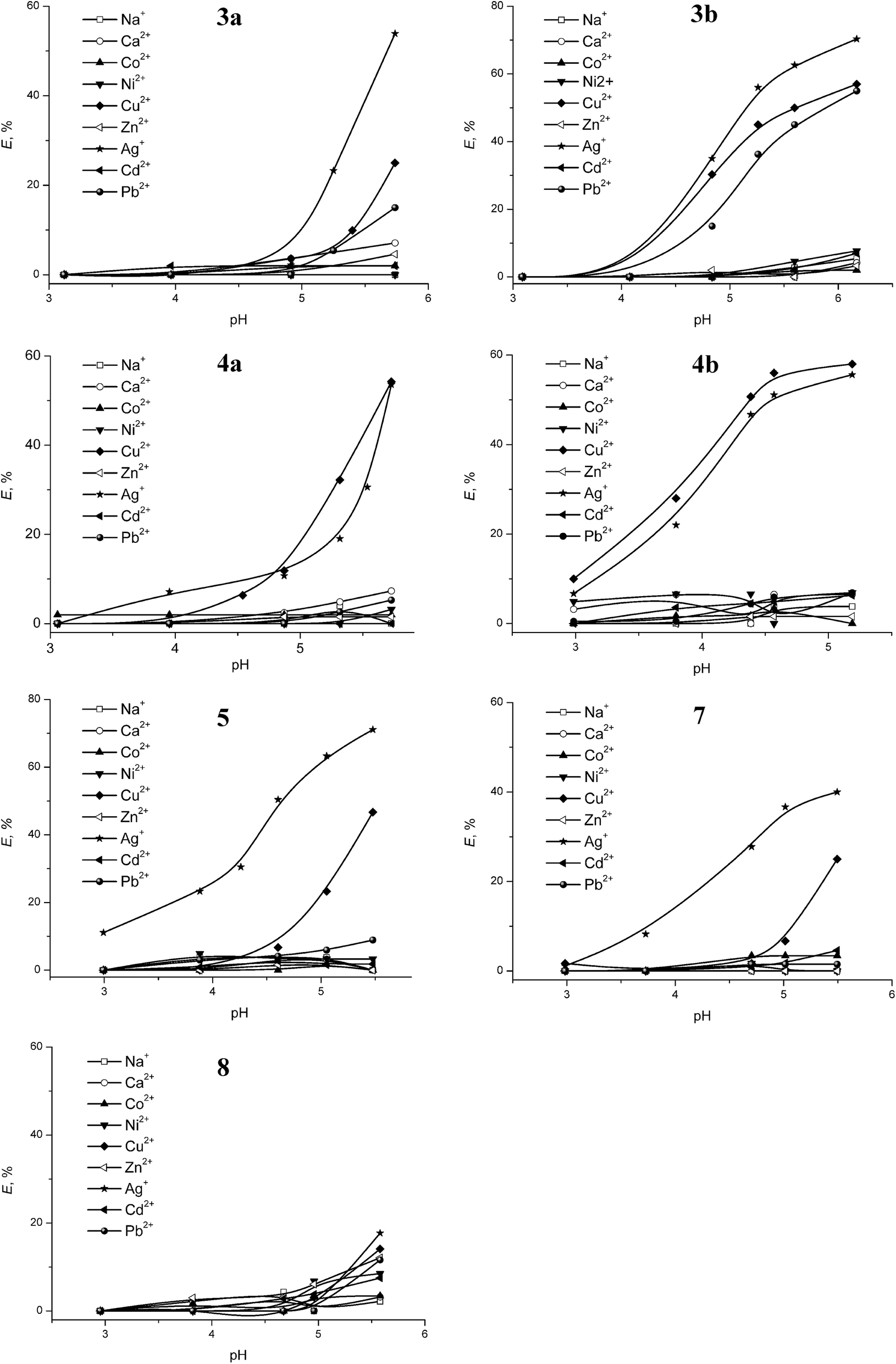 | ||
| Fig. 5 Effect of pH on the extraction percentage (E%) for different metal ions recovered by the ligands 3–7. [L] = 0.2 mM, [metal ion] = 0.1 mM, shaking time = 2 h at 298 K, phase ratio 1:1 (v/v), pH was adjusted with HNO3 and Tris buffer. | ||
It was established that an increase of pH of the aqueous phase up to pH ∼ 5–6 leads to the enhancement of extraction ability of the investigated compounds towards metal ions. This phenomenon obviously may be explained by the ionization of carboxylic groups of the compounds at high pH, except the compound 7. From the other hand, a presence of lone electron pairs at the nitrogen atoms in dihydrazides 4a, 4b, 7 and dihydrazone 5 provides their basic properties. The protonation of hydrazide and hydrazone groups in these compounds, proceeding at the interface in acid conditions, prevents their complex formation with metal cations. This fact seems to be a main reason of the pH depending extraction revealed by the compound 7 having an incapable of deprotonation ester groups instead of the carboxylic ones.
All tetrathiacalix[4]arene derivatives 3–7 and the compound 8 have demonstrated a poor extraction towards s-elements (E < 5% for Na+ and E < 5% for Ca2+). At the same time, the tetrathiacalix[4]arene derivatives recovered selectively certain d- and p- ions at pH ∼ 5–6. The selectivity of the ligands 3a and 3b towards the metal ions is reduced in the row Ag+ > Cu2+ > Pb2+.
The extraction efficiency goes up after removing tert-butyl groups from the upper rim of 1,3-alternate tetrathiacalix[4]arene on going from 3a to 3b: 54% and 70% for Ag+, 25% and 57% for Cu2+, 15% and 45% for Pb2+. In spite of decreasing the lipophilicity of the molecules, the weakening of steric difficulties obviously facilitates a process of metals recovery by the compound 3b. It should be noted that the monomeric carboxylic acid 8 can also recover the Ag+ (18%), Cu2+ (14%) and Pb2+ (11%) metal ions from aqua solutions, but the efficiency and selectivity are substantially lower than for tetrathiacalix[4]arene derivatives. It is worth noting that in the case of carboxylic derivatives of calix[4]-, [5]- and [6]arenes, a more effective binding of Pb2+ relatively Cu2+ has been established earlier.40–44
A replacement of ester groups by the hydrazide ones for 4a and 4b leads to the dramatic decrease of the extraction efficiency towards Pb2+ ion (up to 5–7%). At the same time, the efficiency of the Ag+ and Cu2+ ions recovery by these compounds remains the same. It is known that calix[n]arenes may exhibit an allosteric effect,22,23,45 which was previously detected for the binding of Pb2+ ions by the calix[5,6]arene carboxylic derivatives.43,46 In the case of tetrathiacalix[4]arenes 4a and 4b, the replacement of ester groups on the hydrazide ones can influence on the preorganization of carboxylic binding center similar to the allosteric effect. The hydrazide groups in the calix[4]arenes 4a, 4b and 7, in contrast to the ester ones in the compounds 3a and 3b, are connected by the intramolecular hydrogen bonds (Fig. 2). Such circumstance obviously influences not only on the geometrical parameters, but on the rigidity of the molecules as well. For the dihydrazide derivatives 4a and 4b, these factors lead to a negative effect in the binding of Pb2+ ion which has greater ionic radius (1.19 Å) than Ag+ (0.67 Å) and Cu2+ (0.73 Å) ions.47
An enhanced efficiency of Ag+ (71%) and Cu2+ (47%) ions recovery is observed for the hydrazone 5 in comparison with 3a and 4a. This fact is apparently caused by the simultaneous cooperative participation of the carboxylic and hydrazone binding centers in the coordination of these cations.
In the case of dihydrazide 7 which does not have the carboxylic groups in its structure, a noticeable extraction of Ag+ (40%) and Cu2+ (25%) cations is also observed. It was previously shown by the picrate liquid extraction method that 1,3-alternate tetrahydrazide of p-tert-buthyl-tetrathiacalix[4]arene has revealed a high extraction efficiency exactly for these metal ions, whereas its de-tert-butyl analogue extracted more effectively in the row Ag+ > Ni2+ > Cu2+ > Cd2+.15 Generally, the data obtained for the selectivity of dihydrazide 7 are in accordance with the previously obtained results.
It should be noticed that the recovery of metal cations by the compound 7 can proceed only with participation of a counter-ion, but in the case of the compounds 3a, 3b, 4a, 4b, 5 and 8, the extraction is probably realized via the ion-exchange process. To support this assumption we have performed an extraction experiment in the presence of Pic− anion in aqua phase at pH = 6 ± 0.1 (Fig. 6).
 | ||
| Fig. 6 Extraction percentage (E%) for cations and Pic− as a function of the nature of ligands 3–7. [L] = 0.2 mM, [Metal ion] = 0.1 mM, [HPic] = 0.4 mM, shaking time = 2 h at 298 K, phase ratio = 1:1 (v/v), pH = 6 ± 0.1 was adjusted with HNO3 and Tris buffer. | ||
Picrate is known to form preferentially second sphere complexes and can easier than “hard anions” be transferred from aqua to organic phase due to its enhanced lipophilic properties.48 A transfer of this anion can be readily detected by UV-Vis spectrophotometric method. The analysis of the obtained UV data testified that the extraction of metal cations by the dihydrazide 7 proceeds with the participation of picrate anion (Fig. 6). In the case of dihydrazone 5, a transfer of metal ions is also accompanied by the transfer of Pic− anion. A mixed-mode extraction involving both the ion-exchange process as well as the metal salt transfer is evidently realized for 5. A total extraction percentage of the Ag+ and Cu2+ ions recovery by the compound 5 amounts to 136%. These facts indicate apparently that compound 5 coordinates the metal ions during extraction by both sides of tetrathiacalix[4]arene platform.
Thereby, it can be concluded that the synthesized bi-functional derivatives 3–7 are capable of an effective recovering of the Ag+ and Cu2+ ions from aqua solutions as well as the Pb2+ ions in the case of the compounds 3a and 3b. The extraction of metal cations by the compounds 3–5 is mainly provided due to the binding of the metals by means of carboxylic groups. The preorganization of binding centers on the macrocyclic platform dramatically affects the efficiency and selectivity of the extraction. Moreover, the influence of the substitutes located on opposite side of the macrocycle in 1,3-alternate isomer and non-participated in the binding of the metal cations on the extraction properties has been also revealed. This phenomenon is obviously connected with the allosteric effect realized for this class of macrocyclic compounds.22,23,49
3. Conclusions
In summary, we have reported the synthetic strategy for obtaining of novel bifunctional derivatives of 1,3-alternate tetrathiacalix[4]arene functionalized by carboxylic, ester, hydrazide and hydrazone groups immobilized on opposite sides of the macrocyclic platform. The detailed spectroscopic and structural characteristics as well as receptor properties towards metal ions for the synthesized compounds were established by using X-ray analysis, NMR, IR spectroscopy and liquid–liquid extraction method.The structure of hydrazide fragments in derivatives 4a, 4b and 7 in solution as well as in solid state is characterized by trans-amide conformation which obviously promotes an effective chelate binding of metal ions. The acetylhydrazone fragments in tetrathiacalix[4]arene 5 adopt only the ECN configuration. The ECN/ZCN isomerization was not observed for this compound even at UV-light irradiation of its CDCl3 solutions. At the same time the acetylhydrazone substitutes are in cis- and trans-amide conformations, that leads to the realization of a large number of spatial forms of dihydrazone 5 and as a consequence to the significant complication of NMR and IR spectra. The presence of carboxylic and hydrazide groups in the compounds 4a and 4b predisposed to the hydrogen bond formation does not produce a noticeable amount of associated molecules (∼5%) in CDCl3–CD3OD solutions.
The solvent extraction experiments have demonstrated that an increase of pH of the aqueous phase up to pH ∼ 5–6 leads to the rise of the extraction yields of the metals recovery by the compounds 3a, 3b, 4a, 4b, 5 due to the deprotonation of carboxylic groups. The hydrazide derivative 7 also demonstrates the pH depending extraction properties caused by the protonation of the nitrogen atoms in acid conditions.
The synthesized bifunctional derivatives 3–7 are capable of an effective recognition at pH ∼ 5–6 of the Ag+ and Cu2+ ions from aqua solutions as well as the Pb2+ ions in the case of the compounds 3a and 3b. A nature of the binding centers and their preorganization on macrocyclic platform dramatically affect the efficiency and selectivity of the extraction. The extraction ability of dicarbonic acids 3a and 3b is significantly higher than for monomeric counterpart 8. The extraction efficiency goes up after removing tert-butyl groups from the upper rim of 1,3-alternate tetrathiacalix[4]arene platform on going from 3a to 3b. A replacement of ester groups by the hydrazide and pyridinyl hydrazone ones (compounds 4a,b and 5) leads to the more selective recovery of Ag+ and Cu2+ ions. In the case of dihydrazone 5, a mixed-mode extraction is realized which involves both the ion-exchange process as well as the metal salt transfer.
Thus, we can conclude that the developed synthetic strategy can be successfully applied for the preparing a wide range of bifunctional tetrathiacalix[4]arene derivatives capable of exhibiting excellent receptor properties.
4. Experimental section
4.1. General remarks
All chemicals were used as commercially received without further purification. CHCl3 and DMF were distilled over P2O5. THF was purified by distillation over KOH and sodium. CDCl3 (99.8% isotopic purity) and DMSO-d6 (99.5% isotopic purity) from Aldrich were used for NMR spectroscopy.Microanalyses of C, H, and N were carried out with use of the CHN-3 analyser. Melting points of compounds were measured using a Boetius hotstage apparatus. The purity of the compounds was monitored by TLC. IR absorption spectra were recorded on a Vector-22 Bruker FT-IR spectrophotometer with the resolution of 4 cm−1 as Nujol emulsions and KBr pellets of compounds. Mass spectra (MALDI) were detected on a Finnigan MALDI-TOF Dynamo mass spectrometer. NMR experiments were performed on a Bruker AVANCE-600 spectrometer at 303 K equipped with a 5 mm broadband probehead working at 600.13 MHz in 1H and 150.864 MHz in 13C experiments. Chemical shifts in 1H and 13C spectra (Table 2) were reported relative to the solvent as internal standard (CDCl3δ(1H) = 7.27 ppm, δ(13C) = 77.2 ppm; DMSO δ(1H) = 2.50 ppm, δ(13C) = 39.5 ppm). Assignment was accomplished by means of 2D 1H-13C HSQC and 2D 1H-13C HMBC methods. The pulse programs of the HSQC, HMBC, FT-PGSE and NOESY experiments were taken from Bruker software library. The Fourier transform pulsed-gradient spin-echo (FT-PGSE) experiments50 were performed by BPP-STE-LED (bipolar pulse pair-stimulated echo-longitudinal eddy current delay) sequence.51
4.2. Synthesis
The synthetic routes and the structural formulae of the investigated compounds are shown in Fig. 1. The parent tetrathiacalix[4]arenes 1a52 and 1b53 as well as the tetrathiacalix[4]arenyloxyacetic acid ethyl esters in 1,3-alternate conformation 2a21 and 2b22 were prepared according to the literature methods. The dicarbonic acids 3a and 3b were obtained by the selective hydrolysis of tetrathiacalix[4]arene tetraesters 2a and 2b in the presence of an excess of Cs2CO3 as described by us earlier.241H, 13C NMR and IR data of the synthesized compounds are presented in Tables 1–34.3. Crystal structure
The X-ray diffraction data for the crystals of 4a, 4a·2NH2NH2, 4b·NH2NH2, 5 and 7 were collected on a Bruker Smart Apex II CCD diffractometer in the ω and φ-scan modes using graphite monochromated Mo Kα (λ = 0.71073 Å) radiation at 150(2) K (4a) and at 296(2) K for other samples. Data were corrected for the absorption effect using SADABS program.55 The structures were solved by direct method and refined by the full matrix least-squares using SHELXTL56 and WinGX57 programs. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were inserted at calculated positions and refined as riding atoms except the hydrogen atoms on solvents molecules, hydroxyl groups and amino groups, which were located from difference maps and refined using a riding model. Data collections: images were indexed, integrates, and scaled using the APEX2 (ref. 58) data reduction package. Analysis of the intermolecular interactions was performed using the program PLATON.59 Mercury program package60 was used for figures preparation.Crystallographic data (excluding structure factors) for the structure 4a, 4a·2NH2NH2, 4b·NH2NH2, 5 and 7 have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 1437363–1437367 respectively.
104 reflections measured, 15532 independent reflections (Rint = 0.1253), 797 parameters, 447 restraints. Final indices: R1 = 0.1075, wR2 = 0.2694 (4474 reflections with I > 2σI), R1 = 0.2629 (all data), wR2 = 0.3323 (all data), GoF = 0.875, largest difference in peak and hole (0.545 and −0.533 e Å−3).
326(4) Å3, space group C2/c, Z = 8, μ(Mo Kα) = 0.226 mm−1, ρcalc = 1.226 g cm−3, F(000) = 4448, 13275 independent reflections, 3458 reflections with I > 2σI, 650 parameters, 121 restraints. Final indices: R1 = 0.0952 (I > 2σI), wR2 = 0.2080 (I > 2σI), R1 = 0.2839 (all data), wR2 = 0.2684 (all data), GoF (goodness-of-fit on F2) = 0.763, largest difference in peak and hole (0.562 and −0.481 e Å−3).
221 reflections measured, 9032 independent reflections (Rint = 0.0261), 7845 reflections with I > 2σI, 522 parameters, 38 restraints. Final indices: R1 = 0.0589 (I > 2σI), wR2 = 0.1455 (I > 2σI), R1 = 0.0669 (all data), wR2 = 0.1503 (all data), GoF = 1.110, largest difference in peak and hole (0.944 and −0.620 e Å−3).
, Z = 2, μ(Mo Kα) = 0.204 mm−1, ρcalc = 1.207 g cm−3, F(000) = 1260, 39008 reflections measured, 16924 independent reflections (Rint = 0.2261), 3408 reflections with I > 2σI, 752 parameters, 690 restraints. Final indices: R1 = 0.1530 (I > 2σI), wR2 = 0.4178 (I > 2σI), R1 = 0.3498 (all data), wR2 = 0.4690 (all data), GoF = 0.897, largest difference in peak and hole (0.692 and −0.483 e Å−3).
387 reflections measured, 9433 independent reflections (Rint = 0.0689), 4234 reflections with I > 2σI, 618 parameters, 354 restraints. Final indices: R1 = 0.0606 (I > 2σI), wR2 = 0.1482 (I > 2σI), R1 = 0.1632 (all data), wR2 = 0.1905 (all data), GoF = 1.010, largest difference in peak and hole (0.622 and −0.342 e Å−3).
4.4. Competitive extraction of metal cations
The CHCl3 was saturated with H2O to prevent volume changes during the extraction. Aqueous solutions (10 ml) containing metal salts (NaNO3; Ca(NO3)2·4H2O, Co(NO3)2·6H2O, Ni(NO3)2·6H2O, Cu2(CH3COO)4·2H2O, Zn(NO3)2·4H2O, Pb(NO3)2, AgNO3 and Cd(CH3COO)2·2H2O) were prepared with the concentration for each metal ion 1 × 10−4 M. To adjust pH of the solutions a tris(hydroxymethyl)aminomethane (Tris) and HNO3 were added arbitrarily. The equal volumes of the aqueous mixture of metal salts and the CHCl3 solution of extractant (CL3a,L3b,L4a,L4b,L5,L7,L8 = 2 × 10−4 M) were magnetically stirred in a flask. The extraction equilibrium was reached after vigorous stirring for 1.5 h at 25 °C. Then two phases were allowed to settle for 1 h and afterwards separated by centrifugation.The pH and concentration of metal ions in aqueous phase before and after extraction were measured. The relative concentrations of the cations in the aqueous phase were determined by the applying of atomic absorbance spectrometer AAS 1 N (Carl Zeiss Jena) with the use of oxidative air-acetylene flame. Quantification was made by referring on a standard solution containing a mixture of salts. Blank experiments without added hosts were carried out under the same experimental conditions. The percentage of extraction was calculated as a ratio E% = 100 × (C0 − C1)/C0, where C0 and C1 are the initial and equilibrium concentrations of metal ion in the aqueous solution determined before and after extraction, respectively. E% uncertainties are generally ≤2%.
Notes and references
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, Chichester, UK, 2000 Search PubMed.
- H. W. Lin, Angew. Chem., 2007, 119, 911–914 CrossRef.
- L. Prodi, F. Bolletta, M. Montaliti and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59–83 CrossRef CAS.
- V. M. Mirsky and A. K. Yatsimirsky, Artificial Receptors for Chemical Sensors, Wiley-VCH, Weinheim, 2010 Search PubMed.
- G. Yu, C. Han, Z. Zhang, J. Chen, X. Yan, B. Zheng, S. Liu and F. Huang, J. Am. Chem. Soc., 2012, 134, 8711–8717 CrossRef CAS PubMed.
- G. Yu, M. Xue, Z. Zhang, J. Li, C. Han and F. Huang, J. Am. Chem. Soc., 2012, 134, 3248–13251 Search PubMed.
- G. Yu, X. Zhou, Z. Zhang, C. Han, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2012, 134, 19489–19497 CrossRef CAS PubMed.
- C. D. Gutsche, Calixarenes: An Introduction, The Royal Society of Chemistry, Cambridge, 2nd edn, 2008 Search PubMed.
- Z. Asfary, V. Böhmer, J. Harrowfield and J. Vicens, Calixarenes 2001, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2001 Search PubMed.
- O. K. Sung and C. N. Kye, Bull. Korean Chem. Soc., 2002, 23, 640–642 CrossRef.
- K. Manoj, D. Abhimanew and B. Vandana, Tetrahedron, 2009, 65, 7510–7515 CrossRef.
- X.-I. Ni, H. Tomiyasu, T. Shimizu, C. Pérez-Casas, Z. Xi and T. Yamato, J. Inclusion Phenom. Macrocyclic Chem., 2010, 68, 99–108 CrossRef CAS.
- J. Mendez-Arroyo, J. Barroso-Flores, A. M. Lifschitz, A. A. Sarjeant, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 10340–10348 CrossRef CAS PubMed.
- S. N. Podyachev, S. N. Sudakova, V. V. Syakaev, A. K. Galiev, R. R. Shagidullin and A. I. Konovalov, Supramol. Chem., 2008, 20, 479–486 CrossRef CAS.
- S. N. Podyachev, N. E. Burmakina, S. N. Sudakova, V. V. Syakaev and A. I. Konovalov, Supramol. Chem., 2010, 22, 339–346 CrossRef CAS.
- F. Botha, J. Budka, V. Eigner, O. Hudeček, L. Vrzal, I. Císařová and P. Lhoták, Tetrahedron, 2014, 70, 477–483 CrossRef CAS.
- L. Kovbasyuk and R. Krämer, Chem. Rev., 2004, 104, 3161–3187 CrossRef CAS PubMed.
- S. Patai, The Chemistry of Carboxylic Acids and Esters, London, 1969 Search PubMed.
- S. Patai, The Chemistry of Acid Derivatives, New York, 1979 Search PubMed.
- N. Iki, F. Narumi, T. Fujimoto, N. Morohashi and S. Miyano, J. Chem. Soc., Perkin Trans. 2, 1998, 2745–2750 RSC.
- P. Lhotak and J. Sykora, Collect. Czech. Chem. Commun., 2000, 65, 757–771 CrossRef CAS.
- A. Ikeda, Y. Suzuki, M. Yoshimura and S. Shinkai, Tetrahedron, 1998, 54, 2497–2508 CrossRef CAS.
- J. Budka, P. Lhotak, V. Michlova and I. Stibor, Tetrahedron Lett., 2001, 42, 1583–1586 CrossRef CAS.
- S. N. Podyachev, S. N. Sudakova, B. M. Gabidullin, V. V. Syakaev, A. T. Gubaidullin, W. Dehaen and A. I. Konovalov, Tetrahedron Lett., 2012, 53, 3135–3139 CrossRef CAS.
- S. N. Podyachev, V. V. Syakaev, S. N. Sudakova, R. R. Shagidullin, D. V. Osyanina, L. V. Avvakumova, B. I. Buzykin, Sh. K. Latypov, V. D. Habicher and A. I. Konovalov, J. Inclusion Phenom., 2007, 58, 55–61 CrossRef CAS.
- E. Quinlan, S. E. Matthews and T. Gunnlaugsson, J. Org. Chem., 2007, 72, 7497–7503 CrossRef CAS PubMed.
- M. A. Qazi, Ü. Ocak, M. Ocak, S. Memon and I. B. Solangi, J. Fluoresc., 2013, 23, 575–590 CrossRef CAS PubMed.
- X. Li, S.-L. Gong, W.-P. Yang, Y.-Y. Chen and X.-G. Meng, Tetrahedron, 2008, 64, 6230–6237 CrossRef CAS.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to infrared and Raman spectroscopy, London, Academic Press, 1964 Search PubMed.
- L. J. Bellamy, The IR Spectra of Complex Organic Molecules, London: Methuen, N.-Y, Wiley, 2nd edn, 1958 Search PubMed.
- S. N. Podyachev, N. E. Burmakina, V. V. Syakaev, S. N. Sudakova, R. R. Shagidullin and A. I. Konovalov, Tetrahedron, 2009, 65, 408–417 CrossRef CAS.
- S. N. Podyachev, B. M. Gabidullin, V. V. Syakaev, S. N. Sudakova, A. T. Gubaidullin, W. D. Habicher and A. I. Konovalov, J. Mol. Struct., 2011, 1001, 125–133 CrossRef CAS.
- C. Jaime, J. de Mendoza, P. Prados, P. Nieto and C. Sanchez, J. Org. Chem., 1991, 56, 3372–3376 CrossRef CAS.
- P. Lhotak, Eur. J. Org. Chem., 2004, 1675–1692 CrossRef CAS.
- V. V. Syakaev, S. N. Podyachev, B. I. Buzykin, S. K. Latypov, V. D. Habicher and A. I. Konovalov, J. Mol. Struct., 2006, 788, 55–62 CrossRef CAS.
- V. V. Syakaev, S. N. Podyachev, A. T. Gubaidullin, S. N. Sudakova and A. I. Konovalov, J. Mol. Struct., 2008, 885, 111–121 CrossRef CAS.
- P. Stilbs, Prog. Nucl. Magn. Reson. Spectrosc., 1987, 19, 1–45 CrossRef CAS.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed. Engl., 2005, 44, 520–554 CrossRef CAS PubMed.
- T. Brand, E. J. Cabrita and S. Berger, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 159–196 CrossRef CAS.
- N. T. K. Dung and R. Ludwig, New J. Chem., 1999, 23, 603–607 RSC.
- K. Ohto, Y. Fujimoto and K. Inoue, Anal. Chim. Acta, 1999, 387, 61–69 CrossRef CAS.
- B. B. Adhikari, M. Gurung, H. Kawakita and K. Ohto, Analyst, 2011, 136, 3758–3769 RSC.
- B. B. Adhikari, M. Gurung, A. B. Chetry, H. Kawakita and K. Ohto, RSC Adv., 2013, 3, 25950–25959 RSC.
- B. B. Adhikari, M. Gurung, H. Kawakita and K. Ohto, Solvent Extr. Ion Exch., 2013, 31, 483–498 CrossRef CAS.
- Z. Asfari, V. Lamare, J. F. Dozol and J. Vicens, Tetrahedron Lett., 1999, 40, 691 CrossRef CAS.
- B. B. Adhikari, K. Ohto, M. Gurung and H. Kawakita, Tetrahedron Lett., 2010, 51, 3481–3485 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- Y. Takeda, F. Vögtle and E. Weber, Topics of curr. Chem., Springer, Berlin, 1984, vol. 121, pp. 1–38 Search PubMed.
- Z. Asfari, V. Lamare, J. F. Dozol and J. Vicens, Tetrahedron Lett., 1999, 40, 691–694 CrossRef CAS.
- W. S. Price, NMR Studies of Translational Motion, University Press, Cambridge, 2009 Search PubMed.
- D. Wu, A. Chen and C. S. Johnson, J. Magn. Reson., 1995, 115, 260–264 CrossRef CAS.
- H. Kumagai, M. Hasegawa, S. Miyanari, Y. Sugawa, Y. Sato, T. Hori, S. Ueda, H. Kamiyama and S. Miyano, Tetrahedron Lett., 1997, 38, 3971–3972 CrossRef CAS.
- Y. Higuchi, M. Narita, T. Niimi, N. Ogawa, F. Hamada, H. Kumagai, N. Iki, S. Miyano and C. Kabuto, Tetrahedron, 2000, 56, 4659–4666 CrossRef CAS.
- A. Yamada, T. Murase, K. Kikukawa, T. Arimura and S. Shinkai, J. Chem. Soc., Perkin Trans. 2, 1991, 5, 793–797 RSC.
- G. M. Sheldrick, SADABS, Program for empirical X-ray absorption correction, Bruker-Nonius, 2004 Search PubMed.
- G. M. Sheldrick, SHELXTL v.6.12, Structure Determination Software Suite, Bruker AXS, Madison, Wisconsin, USA, 2000 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- APEX2 (Version 2.1), SAINTPlus, Data Reduction and Correction Program (Version 7.31A), BrukerAXS Inc., Madison, Wisconsin, USA, 2006 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Cryst., 2002, 58, 389–397 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1437363–1437367. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra01730d |
| This journal is © The Royal Society of Chemistry 2016 |