
Influence of over-discharge on the lifetime and performance of LiFePO4/graphite batteries†
Yong Zhenga,
Kun Qianb,
Dan Luob,
Yiyang Lib,
Qingwen Lub,
Baohua Lib,
Yan-Bing He*b,
Xindong Wanga,
Jianling Li*a and
Feiyu Kangb
aState Key Laboratory of Advanced Metallurgy, University of Science and Technology Beijing, Beijing 100083, China. E-mail: lijianling@ustb.edu.cn; Fax: +86 10 62332651; Tel: +86 10 62332651
bEngineering Laboratory for Next Generation Power and Energy Storage Batteries, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, China. E-mail: he.yanbing@sz.tsinghua.edu.cn; Fax: +86 755 26036417; Tel: +86 755 26036417
First published on 2nd March 2016
Abstract
In this study, the degradation of a LiFePO4/graphite battery under an over-discharge process and its effect on further cycling stability are investigated. Batteries are over-discharged to 1.5, 1.0, 0.5 or 0.0 V and then cycled 110 times under over-discharge conditions. The batteries over-discharged to 0.5 and 0.0 V experience serious irreversible capacity losses of 12.56% and 24.88%, respectively. The same batteries lost 7.79 and 24.46% more capacity after they were further subjected to 110 cycles between 3.65 and 2.0 V at 1C/1C, respectively. This shows that a serious loss of active lithium and loss of anode material occur at 0.0 V during both over-discharging and the normal cycling stage. Dissolution and breakdown of solid electrolyte interphase (SEI) films are suggested to be the main reason for degradation under over-discharge at low voltage and further lead to a poor cycling performance. Gas generation can be found on the cycled batteries below 1 V and the gas mainly contains H2, CH4 and C2H6. The structures of LiFePO4 and graphite materials have almost no change according to the results of XRD tests. Half-cell study suggests that almost no irreversible capacity loss occurs at the LiFePO4 cathode, whereas a decline in the capacity is observed at the graphite anode, especially for the batteries over-discharging bellow 1.0 V. Evidence for fierce side reactions at 0.5 and 0.0 V is provided as well, as demonstrated by the developed rich surface chemistry and an significant impedance increase for the aged electrodes.
1. Introduction
Lithium-ion batteries have been attracted a great deal of interest because of their high energy and high power densities. Nowadays they have been widely used as power sources in a wide range of applications ranging from portable electronics (e.g., cellular phones, digital cameras, and laptop computers) to electric transportation [e.g., electric vehicles (EVs) and hybrid electric vehicles (HEVs)]. Lithium iron phosphate (LiFePO4) cathode active material presents a high rate capability and high specific capacity (170 mA h g−1),1 which is excellent in terms of safety, cycle life, and cost.2,3 To date, the advantages of LiFePO4 have make the LiFePO4-based battery play a significant role in the energy storage systems (ESSs) as well as power systems for sustainable vehicles.4–7The safety issue of batteries is still the main concern of both consumers and manufacturers in the applications.8–17 The concern becomes extremely severe especially when the batteries work under abusive conditions such as overcharge, over-discharge, and short current. Over-discharge is one of the factors which can greatly influence the battery performance and safety properties.15,17 Thousands of batteries are connected in series or parallel to form a battery pack to supply the desired power for electric vehicles. The battery pack is working under normal conditions while the battery with the lowest capacity may have been suffering over-discharge. Meanwhile, the cell may cause some serious safety problems because Li-ion battery cells have very low tolerance to abuse when suffer over-discharging. Furthermore, over-discharge may lead to the failure of the whole battery pack or a serious accident. Therefore, the research on decay mechanism under over-discharge of LiFePO4/graphite battery is urgently necessary and important.
Some efforts have been carried out to investigate the over-discharge process. Zhang et al.18 studied the capacity fading mechanism during long-term cycling of over-discharged LiCoO2/mesocarbon microbeads (MCMB) batteries by electrochemical and physical characterization. They proposed that the capacity deterioration of over-discharged battery is mainly caused by the dissolution of copper current collector and the deposition of Cu on the surface of anode during the following charging process. Shu et al.19 researched the over-discharge behaviors of LiFePO4, LiNiO2, and LiMn2O4 materials in different overlithiation voltage limitations. They found that over-discharge can drive irreversible solid-state amorphization process and destroy the electrode structure. Hossain et al.20 compared the over-discharge behavior of Lithium ion batteries with different anodes of MCMB and C–C composite. They proposed that the irreversible capacity loss of C–C composite significantly lower than that of the MCMB carbon and the lithium batteries with C–C composite anode can accept repeated over-discharge without performance deterioration.
From an application standpoint, knowing the influence of over-discharge on the cycling degradation and performance of lithium ion batteries is urgently necessary. However, to our knowledge, for LiFePO4/graphite batteries, there are few reports related to capacity fading mechanism induced by over-discharge especially the normal cycling degradation after over-discharged. Further studies remain necessary to clarify the fading mechanism during over-discharge condition, which can further give substantial reference to the safety design of battery and the build of battery management system (BMS).
In the present work, LiFePO4/graphite batteries were over-discharged for 110 times with 4 different cut-off voltages (1.5, 1.0, 0.5 and 0.0 V, respectively) and then experience normal cycling at 1C/1C for 110 times at 25 °C. The capacity fading mechanism of over-discharge and its effect on the cycling stability is systematically investigated using non-destructive tests combing with post-mortem analysis. An experiment flowchart was created to conduct the over-discharge cycling test. Positive-mortem analysis is employed to investigate the aging mechanism. Based on the test results, over-discharging below 1.0 V may cause a serious deterioration of battery and further lead to an obvious capacity fading under normal cycling.
2. Experimental
2.1 Preparation of power batteries
The pouch LiFePO4/graphite power batteries with a capacity of 650 mA h were prepared, which were nominally 2.4 mm thick, 68 mm wide, and 90 mm long. The batteries used the C–LiFePO4 as cathodes, graphite as anodes, and polyethylene as separator. The LiFePO4 (Aleees, Taiwan) and graphite (Shanshan Technology, China) were used for the cathode and anode materials of the battery. Pouch cells were sealed in an aluminum-laminated polymer film box after the injection of an electrolyte comprising 1 M LiPF6 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of ethylene carbonate (EC), dimethyl carbonate (DMC) and ethylene methyl carbonate (EMC) (1 M LiPF6/EC + DMC + EMC). The cells were firstly cycled with a very low current to form a stable solid electrolyte interface (SEI) layer on the surface of the graphite anode. The normal operating potential range of the batteries are 2.00 V (0% state-of-charge) and 3.65 V (100% state-of-charge).
:1:1 mixture of ethylene carbonate (EC), dimethyl carbonate (DMC) and ethylene methyl carbonate (EMC) (1 M LiPF6/EC + DMC + EMC). The cells were firstly cycled with a very low current to form a stable solid electrolyte interface (SEI) layer on the surface of the graphite anode. The normal operating potential range of the batteries are 2.00 V (0% state-of-charge) and 3.65 V (100% state-of-charge).
2.2 Over-discharge and normal cycling tests
Fig. 1 shows the flowchart for the testing procedure. At each condition, 3 cells were studied in order to avoid errors and to characterize possible discrepancy of behavior. A series of tests were performed according to the testing procedure. These tests were carried out at room temperature (25 °C) as follows: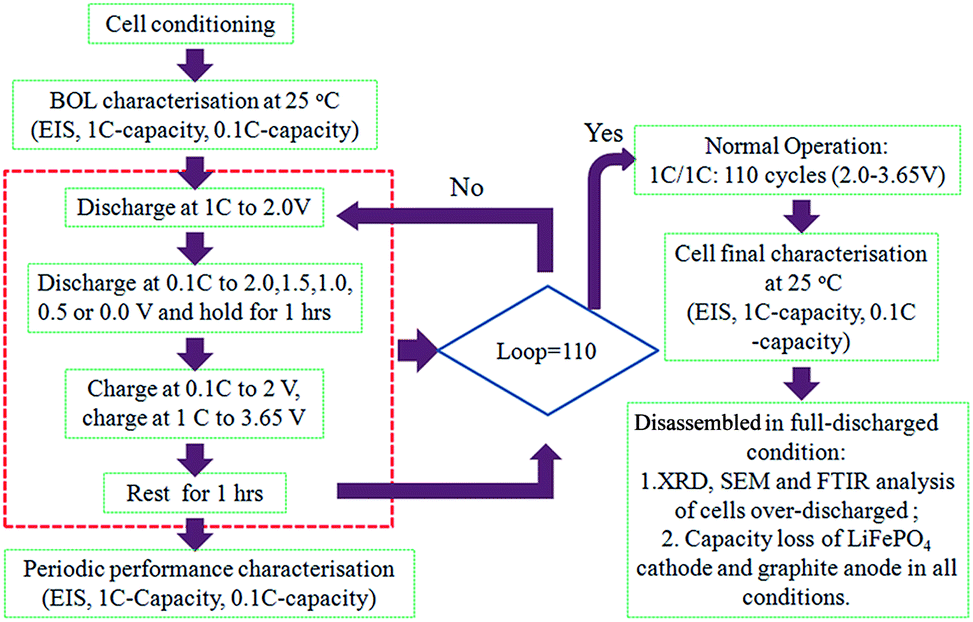 | ||
| Fig. 1 Experiment flowchart of over-discharge measurements. | ||
(1) Cell conditioning, consisted of formation–reactivation cycles carried out as the following procedure: charging the cells with a constant-current/constant-voltage (CC/CV) protocol. The charge rate in the CC stage was 1C, and the cut-off voltage was 3.65 V. The cut-off current was set to 1/50C in the CV stage. Then subsequent discharge at 1C until 2.0 V was reached. Three full charge–discharge cycles were performed.
(2) After cell conditioning step, the initial characterization was carried out, which includes electrochemical impedance spectroscopy (EIS) and capacity measurements. EIS measurement is performed at 60% state of charge (SOC) with a frequency range of 100 kHz to 10 mHz and an AC-oscillation of 5 mV. The capacity at 1C/1C was done using the exact same discharge/charge procedure as that described above for cell conditioning. This is followed by a detailed investigation of the voltage profile, i.e. a capacity test with smaller charge/discharge current (C/10).
(3) After the initial characterization tests were performed, the batteries were cycled 110 times in the following step: discharged to 2.0 V at 1C rate and then discharged further to either of 2.0, 1.5, 1.0, 0.5 or 0.0 V using 0.1C-rate; kept at their pre-fixed discharged voltage for 1 hour; charged at 0.1C to 2 V and then charged to 3.65 V at 1C in CC/CV protocol; 1 hour rest.
(4) A characterization after over-discharge was carried out using the same procedure as in step 2.
(5) Finally, all batteries were cycled 110 times between 3.65 and 2.0 V at 1C/1C. The final characterization tests was then performed using the same procedure as in step 2.
All the characterization tests and cycling tests were completed utilizing Land BT-2013C Battery Test System. The Espec Environmental Chamber was adopted to provide constant temperature environment for all the tests.
2.3 Positive-mortem analysis
The fully discharged batteries before and after over-discharge tests were transferred to a glove box and then dissembled. Before disassembly, the batteries were discharge to 2.0 V at 1C, and then hold the constant voltage of 2.0 V until the discharge current lower than 0.05C. The positive and negative electrodes were washed by dimethyl carbonate (DMC) to remove the electrolyte from the cathode and anode surface. Coin cells were assembled using LiFePO4 and graphite electrodes before and after cycling tests as the cathode, lithium foil as the anode in an argon-filled glove box. The coin cells were examined by Land CT2001A Battery Test System. The capacities of LiFePO4 and graphite electrodes at 0.1C were obtained. EIS tests of coin cells were carried out in the fully discharged state for the coin cells after 4 cycles.Gas-chromatography (GC) was conducted in a PC controlled Agilent 7890A to analyze the gas compositions in batteries. X-ray diffraction (XRD) was carried out on the electrode by a Rigaku D/max 2500/PC diffract meter (Rigaku Corp., Japan) using Cu Kα radiation in an angular range of 10–90° (2θ) with a 0.02° (2θ) step. The morphology and microstructure of the electrodes were observed by using a field emission scanning electron microscope (FE-SEM, HITACH S4800, Japan) with energy dispersive X-ray spectroscopy (EDS). The Fourier transform infrared spectroscopy (FTIR) spectrometer (Bruker VERTEX 70) was applied to analyze the chemical composition of the SEI layer. The electrodes samples for spectroscopy analyses were prepared in the argon-filled glove box in order to avoid reaction with ambient atmosphere.
3. Results and discussion
3.1 Degradation of full batteries during over-discharge and normal cycling conditions
Fig. 2a displays the capacity retention during 110 times of over-discharge with different cut-off voltages. Under normal cycling at 2.0 V, the capacity of the battery was almost no decreased. In contrast, the over-discharged batteries suffered capacity decrease during this period and the cycling behavior of the cell is strongly affected by over-discharge voltage. At reduced discharge voltage, the lifetime of the cell was greatly shortened. Meanwhile, over-discharge to 1.5 and 1.0 V suffered a slightly decrease while that of 0.5 and 0.0 V undergo serious decreased. For the cells over-discharged under the most severe aging conditions (0.0 V), the capacity retention is only about 75.59% relative to the initial capacity. These results indicate that the batteries over-discharging with lower cut-off voltage loss its capacity more quickly. | ||
| Fig. 2 Capacity retention vs. cycle number of LiFePO4/graphite batteries under (a) over-discharged to different voltage, and (b) normal cycling of over-discharged batteries. | ||
The batteries after over-discharge under various conditions were all subjected to 110 cycles between 2.0 and 3.65 V under 1C/1C. The cycling performance is presented in Fig. 2b. After discharged to 2.0 V, 1.5 V and 1.0 V, the cycling capacities fade slowly with cycling number and the capacities losses were 2.02, 2.55, 2.83%, respectively. It is found that the batteries over-discharged to 1.5 and 1.0 V have almost no influence on cycling performance. However, after over-discharged to 0.5 and 0.0 V, it is noteworthy that the batteries show a serious capacity fading of 7.79% and 24.46%, respectively. This result verifies that the batteries over-discharged to 0.5 and 0.0 V subjected serious capacity fading and further caused the poor cycling performance.
In order to investigate the capacity loss under various conditions, the capacity characterization at 1C/1C and 25 °C was performed during an interval of several cycling times. Fig. 3a–d shows the charge and discharge profiles at 1C rate during cycling under 4 different over-discharge conditions. Table 1 presents the capacity loss during over-discharging and normal cycling stage under various conditions. Before cycling, the observed profiles are typical of LiFePO4/graphite full batteries, and the average voltage for discharge was about 3.2 V, which is consistent with previous report.7 Upon over-discharging, the batteries at all conditions undergo capacity fade and the capacity loss enhanced with the over-discharge degree. Noted that after over-discharged at 0.5 and 0.0 V especially for 0.0 V, the charge voltage rises and the discharge voltage decreases, illustrating the incremental of polarization in the full battery system. This may be due to increased resistance in the battery components. In the following normal cycling the batteries over-discharged at 1.5 and 1.0 V experienced a minor capacity losses whereas a significant capacity losses occurred when over-discharged at 0.5 and 0.0 V, especially for 0.0 V. Noted that the voltage plateau remain stable upon cycling at 1.5, 1.0, and 0.5 V while a rise in charge platform and a decline in discharge platform occurred at 0.0 V. Evidently, a serious voltage polarization and capacity loss at 0.0 V indicate that there is an obvious cell deterioration when the battery is over-discharged under this condition.
 | ||
| Fig. 3 Voltage profiles at 1C during cycling under 4 different over-discharge conditions: (a) 1.5 V, (b) 1.0 V, (c) 0.5 V and (d) 0.0 V. Incremental capacity curves of 1/10C discharge profiles at 25 °C for (e) 1.5 V, (f) 1.0 V, (g) 0.5 V and (h) 0.0 V. | ||
| Capacity loss caused by over-discharge/% | Capacity loss during normal cycling after over-discharge/% | |
|---|---|---|
| 2.0 V | 1.08 | 2.02 |
| 1.5 V | 4.11 | 2.55 |
| 1.0 V | 4.86 | 2.83 |
| 0.5 V | 12.56 | 7.79 |
| 0.0 V | 24.88 | 24.46 |
Incremental capacity (IC) is a significant method to identify the active materials loss or lithium loss for capacity degradation during cycling.21,22 Fig. 3e–h shows the IC curves of 1/10C discharge profiles for batteries at over-discharging and normal cycling stage under different conditions. Five peaks can be found from the IC curves. For LiFePO4/graphite battery, in the graphite anode, the lithium intercalation reaction gradually occurs under five distinct staging processes accompanying with the transformation of graphite to Li–graphite intercalation compound (GIC).23,24 Each gives a unique IC peak vs. a reference electrode. In the LiFePO4 cathode, the LiFePO4 electrode associated with the transformation between FePO4 and LiFePO4 usually presents one single flat plateau.25 Therefore the five peaks of LiFePO4/graphite full battery are related to the electrochemical process of graphite electrode.
The peak ① is expected to arise from loss of lithium inventory (LLI) process,22,23,26 which is assigned to the formation of LiC6, i.e. stage 1 Li-GIC.27 The decrease of peak ① indicates that an obvious LLI occurs. As can be seen from Fig. 3e and f, the peaks associated with the highest SOC (peak ①) are significantly decreased at 0.5 and 0.0 V in both over-discharge and further normal cycling stage, indicating that there is an obvious LLI during the two stage under these two conditions. For Fig. 3g and h, noted that the peak ② decreased in the over-discharged stage even though the peak ① in IC curves was not completely lost. It was therefore concluded that, besides LLI, there is also a loss of anode material (LAM),26 which means that the graphite could not be lithiated to the same level as it is initially. All five peaks decreased at 0.0 V under the whole cycling, indicating that a serious LLI and LAM during both over-discharging and normal cycling stage. Meanwhile, all of the peaks of the IC curve at 0.0 V shifted to lower voltages, indicating there may be small increase in the battery resistance during the whole cycling, in agreement with the EIS results in Fig. 4. Besides, it is found that none of the peaks disappeared at all, even though they changed gradually in some cases, so the graphite staging process could apparently be completed upon cycling. From IC curves analysis, we can concluded that there is a significantly LLI and LAM at 0.0 V during both over-discharge and normal cycling stage.
 | ||
| Fig. 4 EIS curves of batteries before and (a) after 110 times of over-discharge to different voltage, (b) after normal cycling stage. (c) The experimental and simulated impedance plots of fresh battery and the equivalent circuit used to fit the EIS. | ||
EIS is a powerful tool that is able to gain insight into the origin of the impedance increase because the various phenomena are separated according to their time constant. Fig. 4a presents the EIS curves of LiFePO4/graphite batteries before and after 110 times of over-discharge with different voltage conditions. We use the equivalent circuit inserted in Fig. 4c to simulate the EIS data by Z-view software. According to the equivalent circuit, the EIS are composed of two partially overlapped semicircles at high to middle frequency and a straight slope line at low frequency end.28 Table 2 shows simulation results. The intersection of the EIS diagram with the real axis refers to the bulk resistance (Rb), which reflected the ohmic resistance of the cell. It may be attributable to electrolyte resistance, electronic resistance of electrode particles and current collectors, and connection resistance between the cell terminals and instrument leads.29 Two depressed semicircles observed at high and mid frequencies were attributed to the resistance (Rsei) and constant phase elements (CPE1) of the solid electrolyte interface (SEI) film, and the charge-transfer resistance (Rct) and constant phase elements (CPE2) of the electrode, respectively. Instead of using the capacitance (Csei) of SEI films and double-layer capacitance (Cdl) of electrodes, CPE1 and CPE2 were used to take the roughness of the electrode surface into consideration. The slope line at low frequencies corresponds to the Warburg impedance (Zw), which was related to lithium ion diffusion within the particles.30
| Resistance | Rb/mΩ | Rsei/mΩ | Rct/mΩ |
|---|---|---|---|
| Fresh | 9.17 | 39.22 | 23.73 |
| 2.0 V | 9.35 | 49.43 | 22.99 |
| 1.5 V | 9.49 | 62.94 | 24.67 |
| 1.0 V | 9.78 | 59.92 | 28.12 |
| 0.5 V | 10.09 | 65.34 | 41.38 |
| 0.0 V | 13.56 | 100.55 | 50.39 |
The extracted parameters of resistances are presented in Table 2. After 110 times of over-discharged, it is found that Rb, Rsei and Rct are all increased and elevated with the increase in over-discharge degree. Rsei increases the most among the three different contributions during over-discharge. Total resistance (Rcell) of the Li-ion cell is mainly contributed by the Rb, Rsei, and Rct. As can be seen from Table 2, a significant increase of Rsei and Rct can be found under deep over-discharging below 1.0 V. After over-discharge at 0.0 V, the Rsei shows an drastically increase of 124.07% relative to the 2 V-cycling. Over-discharging to extremely low voltage can drive the carbon electrode potential to be higher than 3.5 volts.31 Therefore, under a relative low voltage, i.e. lower than 1 V, over-discharging of Li-ion cells may lead to dissolution of the anode SEI-Layer as high negative electrode potentials.32 The SEI film may have been broken up owing to serious de-intercalation and new SEI film may have formed during charging process. This process occurs continuously because of repeated over-discharge to low voltage, i.e. 0.0 V. Furthermore, this may led to gas generation and consequently swelling of the LiFePO4/graphite batteries, as evidenced from the photograph of aged batteries in Fig. 5. A consumption of electrolyte occurred during this process and leads to an increase of Rb. The growing SEI films during over-discharging on the graphite particle surface can lead to the significant increase of Rsei. The Rct increased after over-discharging as well especially under deep discharge, which indicates that the electrochemical reaction process is becoming more difficult. The increased Rct may also attribute to the growing SEI film. Meanwhile, the damage of the anode should occur upon over-discharging due to the dissolution of SEI film, which may also cause the increase of Rct. We can make a conclusion that the dissolution and breakdown of SEI film cause the increase of impedance under deep over-discharging and further lead to a capacity degradation in the batteries, which is consistent with Fig. 2a.
 | ||
| Fig. 5 Pictures of (a) fresh and aged batteries, (b) positive and negative electrodes obtained from fresh and aged batteries, and (c) XRD patterns of LiFePO4 electrodes obtained from area 1 and 2. | ||
Fig. 4b shows the EIS curves of LiFePO4/graphite batteries after normal cycling stage. For LiFePO4/graphite power batteries, kinds of the equivalent circuit models usually employed to analysis EISs30,33–38 (Fig. S2†). For the models30,33–36 in Fig. S2a,† the electrode current could be divided into two parts in parallel: faradic current, which is elicited by the electrochemical reaction; non-faradic current, which is elicited by the charge–discharge of the electrical double layer (CPE). In some other studies,37,38 they considered that the Warburg impedance should be excluded in the parallel circuit, it should be connected with the CPE//Rct unit in series, the equivalent circuits models are shown in Fig. S2b.†
In this paper, the equivalent circuit in Fig. S2a† is used to fit the EIS. In the equivalent circuit, Rct are faradic charge-transfer resistance and the Warburg components represent lithium ion diffusion processes into the electrode matrix and gives rise to a complex impedance, Zw, generally defined according to:35
|
Zw = Rwtanh([jwτw]n)/((jwτ0)n)
| (1) |
The combination of Rct and Zw is called faradic impedance, which reflects kinetics of the cell reactions.33 Low Rct corresponds to a fast kinetics of the faradic reaction. For instance, the Rct value can be used as a kinetic parameter to analyze the corrosion rate of metals.39 There is a close correlation between the Rct and the apparent diffusivity of Li ions in the electrodes.40,41 In addition, as can be seen in Fig. 4c, the experimental and simulated EIS curves were almost identical for the fresh cell, indicative of properly chosen equivalent circuit and the corresponding parameters. Therefore, we use this equivalent circuit to fit the EIS results, which is also can be seen in lots of literatures.30,33–36
The simulation results is presented in Table 3. Noted that a significantly increased of Rsei after normal cycling when the battery over-discharged to 0.5 and 0.0 V. When subjected over-discharged to low voltage of 0.5 and 0.0 V, the SEI film may have been lead to severe damaged owing to dissolution of the anode SEI-Layer. Under further normal cycling stage, the continue growing SEI film should lead to an obvious increase of Rsei. The interfacial reaction owing to the growing SEI film may caused sustainable consumption of active lithium, which result in a poor cycling performance under these two conditions.
| Resistance | Rb/mΩ | Rsei/mΩ | Rct/mΩ |
|---|---|---|---|
| 2.0 V | 9.74 | 58.14 | 21.61 |
| 1.5 V | 9.81 | 59.30 | 24.13 |
| 1.0 V | 10.03 | 65.76 | 28.78 |
| 0.5 V | 10.10 | 78.62 | 39.49 |
| 0.0 V | 14.46 | 121.48 | 53.07 |
3.2 Post-mortem analysis after cycling
In order to further investigate the degradation of active material and confirm the nondestructive electrochemical results, all the aged batteries after over-discharged and further normal cycling were applied for post-mortem analysis. Fig. 5a shows the pictures of full batteries before and after cycling. As can be seen from Fig. 5a, the LiFePO4/graphite batteries at 1.5 and 1.0 V exhibit the same appearance as the fresh battery. Whereas after cycling at 0.5 and 0.0 V, there is some swelling and it seems that a gas generates on the cycled batteries. Furthermore, gas species and ratio of batteries at 0.0 V is investigated and the result is presented in Table 4. It is found that the generated gases mainly contain H2, CH4 and C2H6. The considerable amount of H2 gas at 0.0 V could be attributed to the dissolution and breakdown of SEI films on the graphite anode as high negative electrode potentials. The large amount of CH4 and C2H6 may be originate from interfacial reactions between graphite and surrounding alkyl carbonate solvents. These results provide a direct evidence that the high negative electrode potential at 0.0 V is closely related to the gas generations.| Battery | H2 (%) | N2 (%) | O2 (%) | CO (%) | CH4 (%) | C2H6 (%) | C2H2 (%) | C2H4 (%) | C3H8 (%) |
|---|---|---|---|---|---|---|---|---|---|
| 0.0 V | 35.68 | 2.75 | 0.77 | 0.45 | 53.06 | 6.12 | 0.18 | 0.29 | 0.69 |
The photographs of LiFePO4 cathode and graphite anode before and after cycling are plotted in Fig. 5b. It is found that the morphology of cathode and anode remains almost unchanged at 1.5 and 1.0 V. However, after over-discharging to 0.5 and 0.0 V, we can see the electrode surface of LiFePO4 exhibits grey white in color at several places. From the results of XRD measurements we can see the FePO4 phase enhances obviously in these places. The increased FePO4 phase indicates that there is no enough lithium ions combined with FePO4 to form LiFePO4. These result indicate that an non-uniform active lithium loss occurs on the LiFePO4 when over-discharged below 1 V, especially at 0.0 V. The observed phenomenon may be reveals that the films on the LiFePO4 cathode surface is partially damaged. Meanwhile, it is clearly see that there is a damage on the graphite electrode after over-discharged at 0.5 and 0.0 V. When subjected to over-discharging under these two conditions, the binder may be partially damaged due to the continuous dissolution and reformation of SEI-layer on the graphite particles. This may further caused a partial abscission of graphite particles in some places and a damage of graphite electrodes.
The XRD results of the fully discharged LiFePO4 and graphite electrode dismantled from the fresh and over-discharged batteries are plotted in Fig. 6. As can be seen from Fig. 6a, the XRD peaks of fresh LiFePO4 in the fully discharged state are indexed as an orthorhombic olivine-type structure with a space group of Pnma. It can be seen that the LiFePO4 materials maintain their olivine-type structure after over-discharged at various conditions. Noted that the peak intensity of FePO4 phase enhances obviously after cycling, as we have reported in previous work.42,43 The peak intensity of FePO4 phase increases with the over-discharge degree of battery, suggesting that the FePO4 content increases in aged LiFePO4 electrode. It should be noted that all aged batteries were fully discharged before disassembling for XRD examination. The formation of FePO4 indicates some reversible lithium ion loss in LiFePO4/graphite system during the over-discharge cycling process. Over-discharge does not influence the framework structure of the positive electrode. This result indicates that the capacity loss of battery upon over-discharging is not ascribed to the structure change of the LiFePO4 framework. Moreover, the characteristic peak positions of graphite anode still remained unchanged (Fig. 6b). Thereby, it is concluded that the structure of LiFePO4 and graphite is not the main reason for capacity fading during over-discharge cycling.
 | ||
| Fig. 6 XRD patterns of the (a) LiFePO4 and (b) graphite electrodes obtained from fresh and aged batteries. | ||
SEM was used for spotting possible changes of the electrode morphology upon cycling. The SEM images of LiFePO4 electrodes before and after cycling are plotted in Fig. S1.† The morphology of over-discharged LiFePO4 particle is almost unchanged even though the batteries have been over-discharged for 110 times and further subjected to 110 times of normal cycling. Fig. 7 shows the images of graphite anode between fresh and aged batteries. Before cycling, the graphite particles can be seen clearly with each other. When the batteries were over-discharged to 1.5 and 1.0 V for 100 times and then were charged/discharged for 100 cycles, the morphology of graphite materials has still no obvious difference (Fig. 7c and d). However, for the battery over-discharged to 0.5 V and 0.0 V and cycled for 100 times, the anode surface has some roughening of the particle facets and it seems that a membrane covered on the anode surface at 0.0 V, as seen in Fig. 7e and g and higher magnification of Fig. 7f and h. The apparent roughness of the graphite surface is likely related to a damage of SEI film. This damage may further caused the deterioration of graphite anode. These differences in the morphology of the anode are probably reflected on the impedance profiles during cycling.
 | ||
| Fig. 7 SEM images of graphite electrodes before and after cycling: (a) fresh graphite, and aged at (b) 2 V; (c) 1.5 V; (d) 1 V; (e and f) 0.5 V and (g and h) 0.0 V. | ||
The FTIR measurements of both LiFePO4 and graphite electrodes are presented in Fig. 8. For Fig. 8a, the spectrum of the fresh LiFePO4 electrode displays several peaks at 1140, 1100, 1045, and 950 cm−1. Those peaks around 1200–900 cm−1 can be attributed to P–O bond vibrations.44,45 The bands around 1400 cm−1 corresponding to CO2− stretching vibration46 and the bands at around 550, 578, and 637 cm−1 are related to the bending modes of the PO43− anion.47 The aged electrodes at 2.0, 1.5 and 1.0 V exhibit almost the same spectra as the fresh samples. However, the spectra for aged electrodes at 0.5 and 0.0 V present a new peak at around 1220 cm−1, which are most often related to stretching vibrations from epoxy groups.48 The aged at 0.0 V presents a weak peak at around 1723 cm−1, which can be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.49 Another new peak appears at around 840 cm−1. Such a peak may reflect P–F bonds.50 For the graphite electrode (Fig. 8b), it is found that the IR spectra are clearly influenced by over-discharged at 0.5 and 0.0 V. The spectra of aged electrodes under these two conditions display two new peaks at around 1500 cm−1 and 1420 cm−1, which are probably reflect C–O stretching of Li2CO3 and CO asymmetric stretching of RCOOLi and CH2 bending of (CH2OCO2Li)2,51 respectively. These stuffs could not be clearly identified due to the limited amount of decomposing products. The new peak around 840 cm−1 is attributed to P–F bonds. Those peaks were barely found in the fresh and battery over-discharged at 2.0, 1.5 and 1.0 V, suggesting that the electrodes develop rich surface chemistry upon over-discharging at 0.5 and 0.0 V.
O bonds.49 Another new peak appears at around 840 cm−1. Such a peak may reflect P–F bonds.50 For the graphite electrode (Fig. 8b), it is found that the IR spectra are clearly influenced by over-discharged at 0.5 and 0.0 V. The spectra of aged electrodes under these two conditions display two new peaks at around 1500 cm−1 and 1420 cm−1, which are probably reflect C–O stretching of Li2CO3 and CO asymmetric stretching of RCOOLi and CH2 bending of (CH2OCO2Li)2,51 respectively. These stuffs could not be clearly identified due to the limited amount of decomposing products. The new peak around 840 cm−1 is attributed to P–F bonds. Those peaks were barely found in the fresh and battery over-discharged at 2.0, 1.5 and 1.0 V, suggesting that the electrodes develop rich surface chemistry upon over-discharging at 0.5 and 0.0 V.
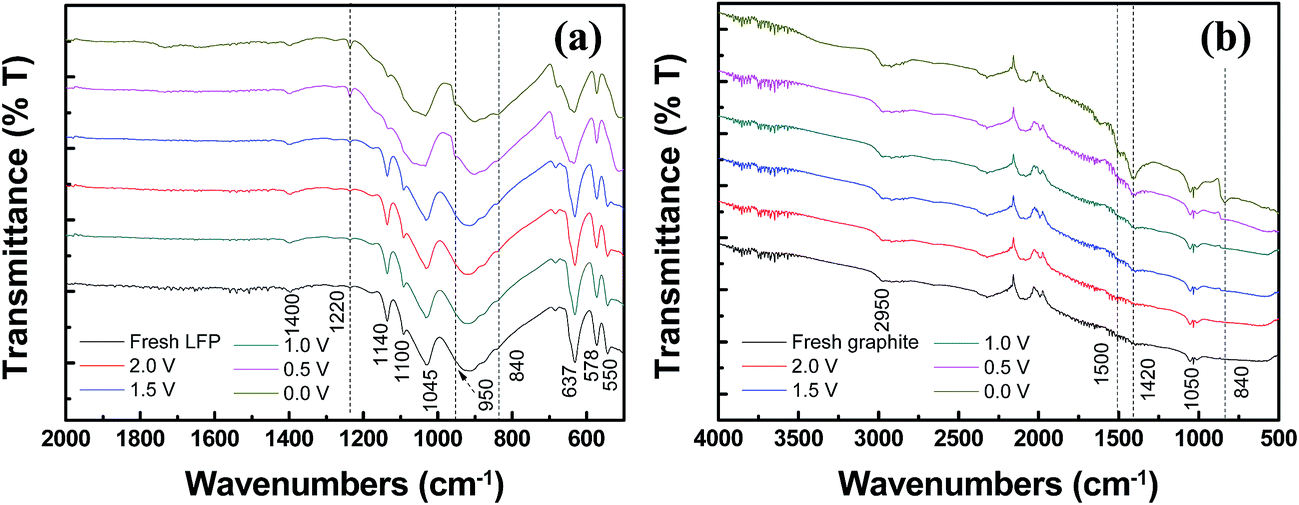 | ||
| Fig. 8 FTIR spectra measured in reflectance mode of fresh and aged electrodes: (a) LiFePO4 cathodes and (b) graphite anodes. | ||
In order to further investigate the fading mechanism during over-discharging, half-cell tests were performed after disassembling the fully discharged full batteries. Fig. 9a presents the charge/discharge curves of Li/LiFePO4 coin cells at 0.1C. It is seen that the capacities are 2.92, 2.87, 2.89, 2.90 and 2.87 mA h for the fresh LiFePO4 and LiFePO4 after over-discharged at 1.5, 1.0, 0.5 and 0.0 V, respectively. The various storage conditions do not lead to significant changes in the capacity values. This result suggests that the electrode material does not undergo any structural degradation that causes capacity fade, which is consistent with the results of XRD results.
 | ||
| Fig. 9 The charge and discharge profiles of coin cells measured at 0.1C and 25 °C both for fresh and aged electrodes: (a) LiFePO4 cathodes and (b) graphite anodes. | ||
Fig. 9b shows the voltage curves of Li/graphite coin cells at 0.1C. It is seen that the capacities are 3.01, 2.87, 2.87, 2.76 and 2.43 mA h for the fresh graphite and graphite after over-discharged at 1.5, 1.0, 0.5 and 0.0 V, respectively. The capacity losses are 4.56%, 4.56% 8.11% and 19.16% for the graphite anodes after over-discharged at 1.5, 1.0, 0.5 and 0.0 V, respectively. A significant change in the graphite capacity can be found for over-discharged at 0.5 and 0.0 V. The serious irreversible capacity decay under these two conditions should be owing to the deterioration of graphite particles resulting from the growing of the SEI films. Therefore, we can conclude that the capacity loss of graphite electrode is a reason for capacity fading under over-discharge conditions.
Fig. 10 displays the electrochemical impedance spectra of fresh and aged electrodes for both LiFePO4 cathode and graphite anode. For both cathode and anode the Nyquist (complex-plane) plot of the impedance has one semicircle in the high frequency region and a straight slope line in the lower frequency region. The size of the semicircles increased upon over-discharge cycling. The dramatic increase of the mid-frequency intercept values suggests that the interfacial resistance of the electrodes rises after cycling. When compared to the EIS results of the graphite electrodes, the absolute values of the LiFePO4 electrodes are relatively lower but the increment ratio in the resistance is higher. For both LiFePO4 and graphite, we can see the size of the semicircle generally increased with the over-discharge degree. It is clearly seen that the highest impedance increase is for the electrodes aged at 0.0 V. The evolution of the interfacial resistance values might be attributed to the increased thickness or lower ion conductivity of the SEI films.
 | ||
| Fig. 10 EIS curves of coin cells for (a) LiFePO4 and (b) graphite electrodes obtained from fresh and cycled batteries. The measurement is performed at 25 °C and in the fully discharged state. | ||
4. Conclusion
This study presents the effect of over-discharge on the degradation and further cyclic stability of LiFePO4/graphite batteries. The extent of capacity fade is found to be directly related to the over-discharge degree. The batteries over-discharging to 0.5 and 0.0 V can leads to serious capacity loss and further lead to a poor cycling performance under normal operating conditions. IC curves indicate that over-discharging to 0.0 V experiences a serious LLI and LAM both in the over-discharging and further normal cycling stage. Dissolution and reformation of SEI film result in the significantly increase of Rsei and caused the serious capacity fading when over-discharged to 0.5 and 0.0 V. XRD measurements indicate that there is no obvious structural change in LiFePO4 and MCMB electrode material upon cycling under various conditions. FTIR results imply that over-discharge to 0.5 and 0.0 V develop rich surface chemistry on both electrodes after cycling. No obvious capacity loss occurs at LiFePO4 cathode according to the half-cell tests. Whereas a serious irreversible capacity loss can be found for graphite anode after cycling, especially at 0.0 V. Evolution of electrode impendence reveals that both cathodes and anodes exhibit increase of impedance and the batteries at 0.0 V experience the highest impedance increase in both electrodes.Acknowledgements
This work was supported by National Science and Technology Support Program (2015BAG01B01), National Key Basic Research Program of China (No. 2014CB932400), National Natural Science Foundation of China (No. 51072131, 51232005 and U1401243), NSAF (No. U1330123), Shenzhen Basic Research Project (No. ZDSYS20140509172959981 and JCYJ20140417115840246), Guangdong Province Innovation R&D Team Plan for Energy and Environmental Materials (No. 2009010025), Production-study-research cooperation project of guangdong province (2014B090901021).References
- S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 2002, 1, 123–128 CrossRef CAS PubMed.
- M. A. Roscher, J. Vetter and D. U. Sauer, J. Power Sources, 2010, 195, 3922–3927 CrossRef CAS.
- H. Joachin, T. D. Kaun, K. Zaghib and J. Prakash, J. Electrochem. Soc., 2009, 156, A401–A406 CrossRef CAS.
- K. Zaghib, M. Dontigny, A. Guerfi, P. Charest, I. Rodrigues, A. Mauger and C. M. Julien, J. Power Sources, 2011, 196, 3949–3954 CrossRef CAS.
- K. Zaghib, P. Charest, A. Guerfi, J. Shim, M. Perrier and K. Striebel, J. Power Sources, 2005, 146, 380–385 CrossRef CAS.
- K. Zaghib, P. Charest, A. Guerfi, J. Shim, M. Perrier and K. Striebel, J. Power Sources, 2004, 134, 124–129 CrossRef CAS.
- J. Shim and K. A. Striebel, J. Power Sources, 2003, 119, 955–958 CrossRef.
- R. Spotnitz and J. Franklin, J. Power Sources, 2003, 113, 81–100 CrossRef CAS.
- W. Cai, H. Wang, H. Maleki, J. Howard and E. Lara-Curzio, J. Power Sources, 2011, 196, 7779–7783 CrossRef CAS.
- Z. Zhang, D. Fouchard and J. R. Rea, J. Power Sources, 1998, 70, 16–20 CrossRef CAS.
- C. K. Lin, Y. Ren, K. Amine, Y. Qin and Z. H. Chen, J. Power Sources, 2013, 230, 32–37 CrossRef CAS.
- J. Lamb, C. J. Orendorff, K. Amine, G. Krumdick, Z. Zhang, L. Zhang and A. S. Gozdz, J. Power Sources, 2014, 247, 1011–1017 CrossRef CAS.
- L. M. Moshurchak, C. Buhrmester, R. L. Wang and J. R. Dahn, Electrochim. Acta, 2007, 52, 3779–3784 CrossRef CAS.
- Q. F. Yuan, F. Zhao, W. Wang, Y. Zhao, Z. Liang and D. Yan, Electrochim. Acta, 2015, 178, 682–688 CrossRef CAS.
- J. Garche, A. Jossen and H. Doring, J. Power Sources, 1997, 67, 201–212 CrossRef CAS.
- H. Yasuhisa and T. Masaru, J. Power Sources, 1997, 67, 349 CrossRef.
- S. Erol, M. E. Orazem and R. P. Muller, J. Power Sources, 2014, 270, 92–100 CrossRef CAS.
- L. L. Zhang, Y. L. Ma, X. Q. Cheng, C. Y. Du, T. Guan, Y. Z. Cui, S. Sun, P. J. Zuo, Y. Z. Gao and G. P. Yin, J. Power Sources, 2015, 293, 1006–1015 CrossRef CAS.
- J. Shu, M. Shui, D. Xu, D. J. Wang, Y. L. Ren and S. Gao, J. Solid State Electrochem., 2012, 16, 819–824 CrossRef CAS.
- S. Hossain, Y. K. Kim, Y. Saleh and R. Loutfy, J. Power Sources, 2003, 114, 264–276 CrossRef CAS.
- M. Dubarry, V. Svoboda, R. Hwu and B. Y. Liaw, J. Power Sources, 2007, 165, 566–572 CrossRef CAS.
- M. Dubarry, B. Y. Liaw, M. S. Chen, S. S. Chyan, K. C. Han, W. T. Sie and S. H. Wu, J. Power Sources, 2011, 196, 3420–3425 CrossRef CAS.
- M. Dubarry and B. Y. Liaw, J. Power Sources, 2009, 194, 541–549 CrossRef CAS.
- M. E. Spahr, T. Palladino, H. Wilhelm, A. Wursig, D. Goers, H. Buqa, M. Holzapfel and P. Novak, J. Electrochem. Soc., 2004, 151, A1383–A1395 CrossRef CAS.
- C. Delacourt, P. Poizot, J. M. Tarascon and C. Masquelier, Nat. Mater., 2005, 4, 254–260 CrossRef CAS.
- M. Dubarry, C. Truchot and B. Y. Liaw, J. Power Sources, 2012, 219, 204–216 CrossRef CAS.
- D. Aurbach, B. Markovsky, I. Weissman, E. Levi and Y. Ein-Eli, Electrochim. Acta, 1999, 45, 67–86 CrossRef CAS.
- Y. B. He, Z. Y. Tang, Q. S. Song, H. Xie, Q. H. Yang, Y. G. Liu and G. W. Ling, J. Power Sources, 2008, 185, 526–533 CrossRef CAS.
- Y. C. Zhang and C. Y. Wang, J. Electrochem. Soc., 2009, 156, A527–A535 CrossRef CAS.
- Y. B. He, M. Liu, Z. D. Huang, B. Zhang, Y. Yu, B. H. Li, F. Y. Kang and J. K. Kim, J. Power Sources, 2013, 239, 269–276 CrossRef CAS.
- Z. Mao, Abs.304, proceedings of the 206th Electrochemistry Society Conference, Honolulu, Hawaii, 2006 Search PubMed.
- C. Kishiyama, M. Nagata, T. Piao, J. Dodd, P.-N. Lam and H. Tsukamoto, Abs. 245, proceedings of the 204th Electrochemistry Society Conference, Orlando FL, 2003 Search PubMed.
- S. S. Zhang, K. Xu and T. R. Jow, Electrochim. Acta, 2004, 49, 1057–1061 CrossRef CAS.
- H. M. Cho, W. S. Choi, J. Y. Go, S. E. Bae and H. C. Shin, J. Power Sources, 2012, 198, 273–280 CrossRef CAS.
- P. S. Attidekou, S. Lambert, M. Armstrong, J. Widmer, K. Scott and P. A. Christensen, J. Power Sources, 2014, 269, 694–703 CrossRef CAS.
- Q. N. Wang, D. L. Chao, W. H. Zhou, Y. G. Chen and C. L. Wu, J. Rare Earths, 2013, 31, 772–777 CrossRef CAS.
- B. Yan, M. S. Li, X. F. Li, Z. M. Bai, L. Dong and D. J. Li, Electrochim. Acta, 2015, 164, 55–61 CrossRef CAS.
- X. F. Li, J. Liu, X. B. Meng, Y. J. Tang, M. N. Banis, J. L. Yang, Y. H. Hu, R. Y. Li, M. Cai and X. L. Sun, J. Power Sources, 2014, 247, 57–69 CrossRef CAS.
- H. Ma, S. Chen, L. Niu, S. Zhao, S. Li and D. Li, J. Appl. Electrochem., 2002, 32, 65–72 CrossRef CAS.
- M. Mohamedi, D. Takahashi, T. Itoh and I. Uchida, Electrochim. Acta, 2002, 47, 3483–3489 CrossRef CAS.
- K. Dokko, M. Mohamedi, M. Umeda and I. Uchida, J. Electrochem. Soc., 2003, 150, A425–A429 CrossRef CAS.
- Y. Zheng, Y. B. He, K. Qian, B. H. Li, X. D. Wang, J. L. Li, S. W. Chiang, C. Miao, F. Y. Kang and J. B. Zhang, Electrochim. Acta, 2015, 176, 270–279 CrossRef CAS.
- Y. Zheng, Y. B. He, K. Qian, B. H. Li, X. D. Wang, J. L. Li, C. Miao and F. Y. Kang, J. Alloys Compd., 2015, 639, 406–414 CrossRef CAS.
- X. H. Guan, C. Shang, J. Zhu and G. H. Chen, J. Colloid Interface Sci., 2006, 293, 296–302 CrossRef CAS PubMed.
- M. Koltypin, D. Aurbach, L. Nazar and B. Ellis, Electrochem. Solid-State Lett., 2007, 10, A40–A44 CrossRef CAS.
- Y. Xu, Y. Lu, L. Yan, Z. Yang and R. Yang, J. Power Sources, 2006, 160, 570–576 CrossRef CAS.
- Z. Zheng, W. K. Pang, X. Tang, D. Jia, Y. Huang and Z. Guo, J. Alloys Compd., 2015, 640, 95–100 CrossRef CAS.
- X. Tong, H. Wang, G. Wang, L. Wan, Z. Ren, J. Bai and J. Bai, J. Solid State Chem., 2011, 184, 982–989 CrossRef CAS.
- Y.-G. Huang, F.-H. Zheng, X.-H. Zhang, Q.-Y. Li and H.-Q. Wang, Electrochim. Acta, 2014, 130, 740–747 CrossRef CAS.
- D. Aurbach, M. Moshkovich, Y. Cohen and A. Schechter, Langmuir, 1999, 15, 2947–2960 CrossRef CAS.
- P. Verma, P. Maire and P. Novak, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01677d |
| This journal is © The Royal Society of Chemistry 2016 |