
Controlling the resolution and duration of pulsatile release from injectable magnetic ‘plum-pudding’ nanocomposite hydrogels†
Abstract
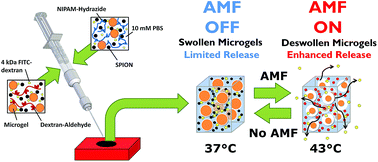
Manipulation of the relative swelling and volume fractions of the microgel and bulk hydrogel phases of nanocomposite hydrogels containing superparamagnetic iron oxide nanoparticles (SPIONs) is demonstrated to enable higher on–off state resolution and enhanced total duration of pulsatile drug release potential when the nanocomposites are activated by an alternating magnetic field. Adjusting the microgel chemistry to create microgels that have less deswelling below 37 °C and more proportional deswelling between 37 °C and 43 °C, increasing the volume fraction of microgels in the nanocomposite (while maintaining the mechanical stability of the nanocomposite), and limiting the swelling capacity of the surrounding hydrogel were all found to improve the degree of enhanced release that occurs after an externally-operated AMF pulse. Collectively, these results serve both to optimize the function of these nanocomposite materials for pulsatile drug delivery as well as confirm the proposed mechanism of pulsatile release, by which free volume is generated within the nanocomposite at the thermoresponsive microgel–bulk hydrogel interface upon internal heating of the device via SPION-driven hysteresis heating of the nanocomposite in an alternating magnetic field. Coupled with the injectability, degradability, mechanical stability, and cytocompatibility of these nanocomposites, we anticipate potential applications as advanced ‘smart’ drug delivery technologies that can be operated via an external and non-invasive trigger.
 Please wait while we load your content...
Please wait while we load your content...

