
Tailoring of ammonia reduced graphene oxide into amine functionalized graphene quantum dots through a Hofmann rearrangement
Abstract
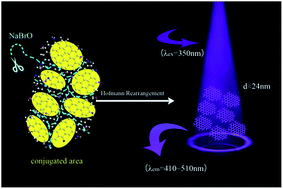
Graphene quantum dots (GQDs) are regarded as promising materials in building biocompatible nanodevices. This paper puts forward a protocol to fabricate amine functionalized graphene quantum dots (afGQDs) by tailoring and exfoliating multilayered ammonia reduced graphene oxide (NH2-G) into afGQDs through a Hofmann rearrangement. The principle on how a Hofmann rearrangement assists in tailoring and exfoliating of NH2-G sheets into afGQDs was posited. The size distribution of afGQDs was tuned by a simple but efficient method based on the adjustment of sodium hypobromite dosage, accompanied by hydrolysis and filtration. The afGQDs emitted broad spectral wavelengths photoluminescence (PL) with two peaks centered at 430 and 510 nm, attributed to unmodified graphene oxide quantum dots and the amine group respectively.
 Please wait while we load your content...
Please wait while we load your content...

