
Preparation of well-defined fibrous hydrogels via electrospinning and in situ “click chemistry”†
Abstract
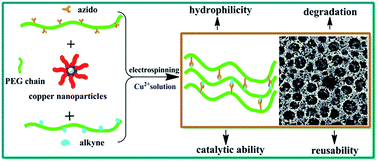
In this work, well-defined PEG-based fibrous hydrogels (FH's) were successfully prepared via electrospinning and in situ copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. Initially, the linear functional PEG derivatives with pendant alkynyl groups (PEGn(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH))m and with azido moieties (PEGn(N3))m were synthesized via epoxide-amine chain-extension reaction between poly(ethylene glycol) diglycidyl ether (PEGDGE) and propargylamine/1-azido-3-aminopropane. Subsequently, the PEG-based FHs were fabricated from the blends of poly(ethylene oxide) (PEO) and the functional PEG derivatives via electrospinning and in situ CuAAC reaction using the encapsulated copper nanoparticles as the catalyst. The blends of PEO and the functional PEG derivatives were also utilized to prepare the microscopic hydrogels (MH's). The properties of the FH's and MH's were investigated by scanning electron microscopy (SEM) observation, swelling ratios, differential scanning calorimetry (DSC) and in vitro degradation. The copper nanoparticles-encapsulated FH's and MH's were further used to catalyze the CuAAC reaction in a small molecule model. The reusability of the FH's for the CuAAC reaction was also studied.
CH))m and with azido moieties (PEGn(N3))m were synthesized via epoxide-amine chain-extension reaction between poly(ethylene glycol) diglycidyl ether (PEGDGE) and propargylamine/1-azido-3-aminopropane. Subsequently, the PEG-based FHs were fabricated from the blends of poly(ethylene oxide) (PEO) and the functional PEG derivatives via electrospinning and in situ CuAAC reaction using the encapsulated copper nanoparticles as the catalyst. The blends of PEO and the functional PEG derivatives were also utilized to prepare the microscopic hydrogels (MH's). The properties of the FH's and MH's were investigated by scanning electron microscopy (SEM) observation, swelling ratios, differential scanning calorimetry (DSC) and in vitro degradation. The copper nanoparticles-encapsulated FH's and MH's were further used to catalyze the CuAAC reaction in a small molecule model. The reusability of the FH's for the CuAAC reaction was also studied.
 Please wait while we load your content...
Please wait while we load your content...

