
Novel glycyrrhetinic acid conjugated pH-sensitive liposomes for the delivery of doxorubicin and its antitumor activities
Qing Chena,
Huaiwei Dingb,
Jinxing Zhoua,
Xiufeng Zhaoc,
Jiulong Zhanga,
Chunrong Yangd,
Kexin Lia,
Mingxi Qiaoa,
Haiyang Hua,
Pingtian Dinga and
Xiuli Zhao*a
aSchool of Pharmacy, Shenyang Pharmaceutical University, Shenyang, 110016, P.R. China. E-mail: raura3687yd@163.com; Fax: +86 24 23986308
bSchool of Pharmaceutical Engineering, Shenyang Pharmaceutical University, 110016, P.R. China
cHongqi Hospital of Mudanjiang Medical University, Mudanjiang, 157000, P.R. China
dCollege Pharmacy of Jiamusi University, 148th Xuefu Street, Jiamusi, Heilongjiang 154007, P.R. China
First published on 5th February 2016
Abstract
Over the last few decades pH-sensitive drug delivery systems have been successfully developed for the treatment of cancers by improving the therapeutical effect. In this study, a novel pH-sensitive conjugate glycyrrhetinic acid–polyethylene glycol–Schiff bond–cholesterol (GPSC) has been synthesized successfully and used to construct doxorubicin (DOX)-loaded liposomes (GPSLP/DOX) with both pH-sensitive features and active targeting ability. DOX was incorporated into liposomes using a thin-film hydration method with a relatively high encapsulation efficiency (EE%) and drug loading content (LC%). Physicochemical characteristics, in vitro release behavior, cellular toxicity and cellular uptake, in vivo biodistribution as well as in vivo antitumor activities of GPSLP/DOX were investigated. GPSLP/DOX showed significantly pH-sensitive features in the in vitro release assay. All the blank liposomes were nontoxic in the in vitro cytotoxicity assay. In the MTT assay, GPSLP/DOX showed the highest cell cytotoxicity among all the groups. A cellular uptake study revealed that GPSLP/DOX could be taken up efficiently via receptor-mediated endocytosis and pH-responsive drug release of DOX into cytoplasm, which resulted in a higher cytotoxicity and therapeutical effect. An in vivo NIR fluorescence image study showed that GPSLP/DOX could specifically accumulate in the liver and tumor sites via receptor mediated endocytosis. In vivo antitumor activity results showed that this novel GPSLP/DOX could significantly inhibit tumor growth and prolong survival time due to its pH-responsive behaviour and GA-mediated endocytosis, resulting in a lower systematic toxicity and higher therapeutical effect. All these results confirm that this GA-mediated pH-sensitive GPSLP/DOX is a novel nanocarrier for the delivery of the antitumor agent DOX to reach higher toxicity effects against tumor tissue.
1 Introduction
Cancer is the leading cause of death with an estimated 8.2 million deaths worldwide.1 Hepatocellular carcinoma (HCC) is the fifth most common cancer in the world and the third most common cause of cancer mortality.2–4 Despite various anticancer strategies being used in clinics, chemotherapy continues to be an important therapeutical option for different malignancies, especially for primary advanced and metastatic tumours.5–7 However, severe side effects and systematic toxicity limit the tolerable dose of many antitumor drugs,8,9 including doxorubicin (DOX). Targeted tumor therapy is one of the most ideal options for patients undergoing chemotherapy as it could increase therapeutical efficacy and reduce adverse effects.10–12 Therefore, it is extremely important to seek new therapeutic strategies to treat liver cancer.In recent years, various drug delivery systems (DDS) have been developed, which could provide targeted delivery anticancer drugs to tumor tissue. Liposomes based delivery system, as an appealing candidate with decreased side effects and enhanced tumor delivery, has attracted considerable attention.13–15 As is known to us all, the membrane of liposome is consist of phosphatidylcholine (PC) and cholesterol. Due to the structure of liposomes is similar to the cell membrane, it is easy to be untaken by cells. However, easily recognized by the reticuloendothelial system (RES) in blood system is becoming the major barrier in clinical application of liposomes.16,17 To solve this problem, polyethylene glycol (PEG) modified liposomes has been widely utilized to reach long circulation and avoid the elimination by the RES.18,19 This long circulating effect enables these small-sized carriers to “passively” accumulate in tumor tissue through the enhanced permeation and retention (EPR) effect. However, the PEG shell of the liposomes seriously hindered the interactions between the DDS and cells membrane after arriving at tumor tissues.20,21 Ideally, a stable PEG coating is required for longer blood circulation to avoid being recognized by the RES. After reaching target tissue, PEG chain becomes unnecessary or even undesirable. Removal of the PEG shielding can allow for more efficient cellular association of the carrier. Furthermore, hydrophilic shell of PEG will make a weak exclusion between liposomes and the cell membrane, thus decrease the cellular uptake and showed lower cytotoxicity.22,23 There are multiple design strategies that can be implemented to overcome the barriers associated with the use of PEGylation, including the conjugation of PEG on the surface of liposomes via pH sensitive,18 matrix metalloprotease (MMP) sensitive,24,25 thermo sensitive26,27 and redox sensitive.28 Among these approaches, pH-sensitive chemical bonds have been most widely used to design sensitive nano-systems for drug delivery in cancer therapy. Tumours have been demonstrated to exhibit acidic pH ranging from 5.7 to 7.0, while the pH of normal tissue is 7.4.29,30 Significant pH difference could also be found at the subcellular level: pH value of early endosomes and late endosomes were 5.5 and 4.5, respectively.31 As a result, the design of pH-sensitive acid-labile liposome may be a good strategy for cancer treatment, owing to the existence of mildly acidic microenvironment in both the interstitium of solid tumours and endosomes of tumor cells. Schiff base bond is an excellent pH-sensitive candidate bond which is easy to construct and can be hydrolyzed by the acidic pH of tumor microenvironment.32,33
In order to ensure the anticancer drug could efficiently accumulate in tumour cells, it is necessary to design an effective delivery system which could not only incorporate the therapeutic agents but could also be efficiently uptaken by the tumour cells. Glycyrrhetinic acid (GA) is one of the main bioactive compounds of licorice and is widely used in medicine for the treatment of much pathology.34–36 Recent reports also showed that carriers modified with GA could reach higher accumulation in the hepatocytes because of abundant GA receptors on hepatocyte membranes, suggesting that vehicles modified with functional group-GA could improve the tumor targeting efficiency for hepatocellular carcinoma chemotherapy.37,38
Here, we report the design of a multifunctional liposomal preparation acting GA as a targeting moiety for enhanced cellular delivery ability and anticancer activity. This drug delivery system was relatively stable in blood system, while degraded in tumour microenvironment via pH-sensitive Schiff base bond between a long shielding PEG chains and cholesterol (PEG2k–SIB–Chol, PSC) (Scheme 1). As another important component of liposome membrane, cholesterol is more chemically stable and easy to be modified compared with phospholipids.39–41 Instead of coupling PEG to phospholipids as described previously,42 we coupled functional PEG via Schiff base linkage to cholesterol as lipidic anchor. This cholesterol–Schiff base–PEG derivative also contained an amino-group moiety to provide the possibility of coupling receptor ligands such as GA. We hypothesized that PEGylated liposomes accumulate in target sites via the GA (“active” targeting). Within the “acidified” of the tumour microenvironment, the carriers will lose their PEG coating by hydrolysis of a Schiff base pH-sensitive bond, and facilitate the tumor cellular uptake. Once internalized in target cells, DOX was expected to release at the acidic pH due to escape from endosomes with the help of the DOPE, allowing cytoplasmic and nuclear accumulation of released DOX.
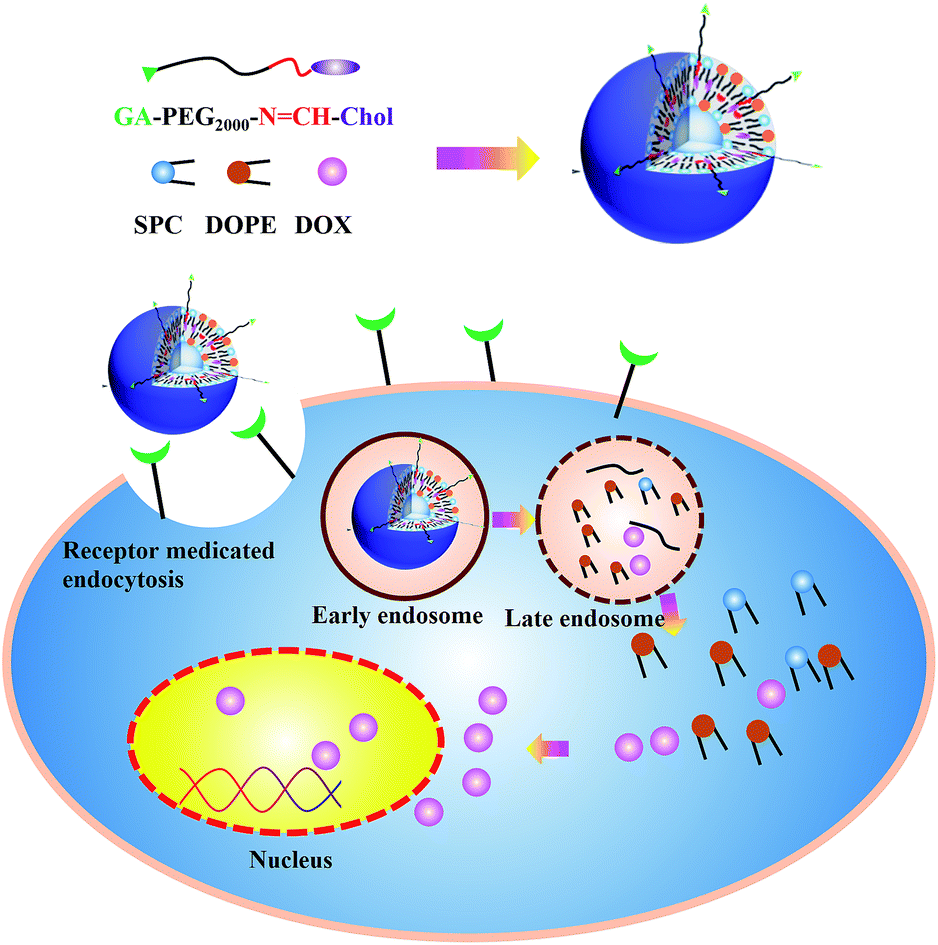 | ||
| Scheme 1 Schematic diagram of GPSLP/DOX and mechanism of GPSLP/DOX to enhance antitumor activities. | ||
2 Materials and methods
2.1. Materials and animals
GA (purity >98% by HPLC) was purchased from Fujie Pharmaceutical Co., Ltd. (Xi'an, China). DOX·HCl was obtained from Huafeng United Technology Co., Ltd. (Beijing, China). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl) and N-hydroxyl-succinimide (NHS) were obtained from GL Biochem., Ltd. Soybean phospholipid (SPC, no. 20140728) was manufactured by Shanghai Advanced Vehicle Technology (AVT) Pharmaceutical Co., Ltd.; cholesterol (Chol, no. 20140908) was purchased from Anhui Tianqi Chemical Technology Co., Ltd.; amino–polyethylene glycol–carboxyl group (NH2–PEG2000–COOH, MW, with 2 kDa of PEG chains) was purchased from Seebio Biotech, Inc. (Shanghai, PR, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was also obtained from Sigma Aldrich. Cholesteryl chloroformate were purchased from J&K Scientific LTD. Dichloromethane (DCM, 99%, kelong chemical, Chengdu, China) was distilled over anhydrous sodium sulfate. Tetrahydrofuran (THF, 99%, Kelong chemical, Chengdu, China) was freshly distilled over sodium prior to use. All other reagents were of commercial special grade and used without further purification.Nude mice (20 ± 2 g) were used in the biodistribution and antitumor assay. All care and handing of animals were performed with the approval of Institutional Authority for Laboratory Animal Care of Shenyang Pharmaceutical University.
2.2. Synthesis and characterization of GA-modified PEG2000–SIB–Chol
To synthesis the GA–polyethylene glycol (PEG)–Schiff base–cholesterol (GPSC) conjugates, para-hydroxyl benzaldehyde and para-phenylenediamine were used to constitute pH sensitive Schiff base bond. The whole process can be divided into two steps:(i) Synthesis of NH2–PEG–SIB–Chol. Cholesteryl chloroform-mate was reacted with para-hydroxyl benzaldehyde (molar ratio = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in dry DCM at room temperature under argon in the presence of N-ethyldiisopropylamine (DIPEA) for about 4 h. After thin layer chromatography (TLC) showed the disappearance of para-hydroxyl benzaldehyde, the reaction mixture was poured into distilled water and used DCM to extract. The extraction was evaporated under vacuum. The residue was purified on a silica-gel chromatography column (DCM:MeOH = 1:1) to get Chol–CHO. Then, Chol–CHO and para-phenylenediamine (molar ratio = 1:2) were reacted in methylbenzene with gentle stirring at 120 °C in oil bath overnight. After TLC showed the disappearance of Chol–CHO, the mixture was evaporated under vacuum, the residue was dissolved in CH2Cl2, and the insoluble material was purified on a silica-gel chromatography column. The collected solution was evaporated under vacuum, the residue (NH2–SIB–Chol) was evaporated again under vacuum and stored under −20 °C until use.
:1) in dry DCM at room temperature under argon in the presence of N-ethyldiisopropylamine (DIPEA) for about 4 h. After thin layer chromatography (TLC) showed the disappearance of para-hydroxyl benzaldehyde, the reaction mixture was poured into distilled water and used DCM to extract. The extraction was evaporated under vacuum. The residue was purified on a silica-gel chromatography column (DCM:MeOH = 1:1) to get Chol–CHO. Then, Chol–CHO and para-phenylenediamine (molar ratio = 1:2) were reacted in methylbenzene with gentle stirring at 120 °C in oil bath overnight. After TLC showed the disappearance of Chol–CHO, the mixture was evaporated under vacuum, the residue was dissolved in CH2Cl2, and the insoluble material was purified on a silica-gel chromatography column. The collected solution was evaporated under vacuum, the residue (NH2–SIB–Chol) was evaporated again under vacuum and stored under −20 °C until use.
For the synthesis of the PEG–SIB–Chol conjugate, COOH–PEG2000–NH2 and NH2–SIB–Chol (molar ratio = 1:1.5) were reacted in DCM with gentle stirring at room temperature in the presence of DIPEA and EDC over night. After TLC showed the disappearance of COOH–PEG2000–NH2, the mixture was extracted by distilled water and DCM. After evaporated the DCM under vacuum. The residue was dissolved in chloroform and filtered again to purify production.
(ii) Synthesis of GA–PEG–SIB–Chol (GPSC). For the synthesis of the GPSC conjugate, PEG–SIB–Chol and GA (molar ratio = 1:1) were reacted in DCM with gentle stirring in the presence of DIPEA, HATU and EDC at room temperature for 48 hours. The GA–PEG–SIB–Chol conjugate was extracted same as the method of PEG–SIB–Chol conjugate.
2.3. Preparation of DOX-loaded liposome
DOX·HCl was stirred with quintuple the number of mole of TEA in 12.5% methanol/chloroform mixture overnight to obtain the DOX base.31 Liposomes were made by the established thin film hydration method followed by extrusion. GA modified PEGylated pH-responsive liposome (GPSLP/DOX) was prepared using thin film hydration method. DOX:SPC:DOPE:Chol:GPSC = 4:40:40:5:5 (total lipid content: 3 mol ml−1) were distilled in ethanol, dried into a thin film on a rotary evaporator, and then hydrated with PBS buffer (pH 7.4), followed by 1 h (37 °C) incubation and 2 min bath-type sonication. Then it was sonicated with a probe-type sonicator at 200 W power for 3 min. The resulting liposomes were filtered through polycarbonate membranes of gradually decreasing pore sizes (0.45 and 0.22 μm). Unloaded DOX was removed using cation exchange resin-mini column centrifugation method. DOX Lip (LP/DOX) was prepared using the same way except GPSC was replaced by cholesterol. The prescription of DOX loaded PEG-Lip (PLP/DOX) was DOX:SPC:DOPE:Chol:mPEG2000–Chol = 4:40:40:5:5 and prepared using the same way. Different blank liposomes were prepared in the same method without adding any DOX.
2.4. Characterization of DOX-loaded liposomes
The particle size distribution and zeta potential of different liposomes were determined. Briefly, different formulations were dissolved in distilled water to reach a proper concentration and detected using a laser light scattering technique (LLS; Nano-2S90 Zetasizer, Malvern, UK).The morphology of the DOX-loaded liposomes were observed using transmission electron microscopy (TEM) (Jeol JEM-2000EX, Tokyo, Japan). In order to avoid multiscattering phenomenon, liposome dispersions were diluted properly with purified water. The samples were negatively stained by 4% phosphotungstic acid and dried on carbon-coated grids for observation.
The drug encapsulation efficiency (EE%) and drug loading content (LC%) of DOX-loaded liposomes were determinated by the cation exchange resin-mini column centrifugation method. DOX was determined using a UV-Vis spectrophotometer at 481 nm. Samples (100 μl) were loaded onto a mini-column (56 mm × 8 mm i.d.) and eluted with DI water to separate unloaded DOX. The EE% and LC% could be calculated as follows:
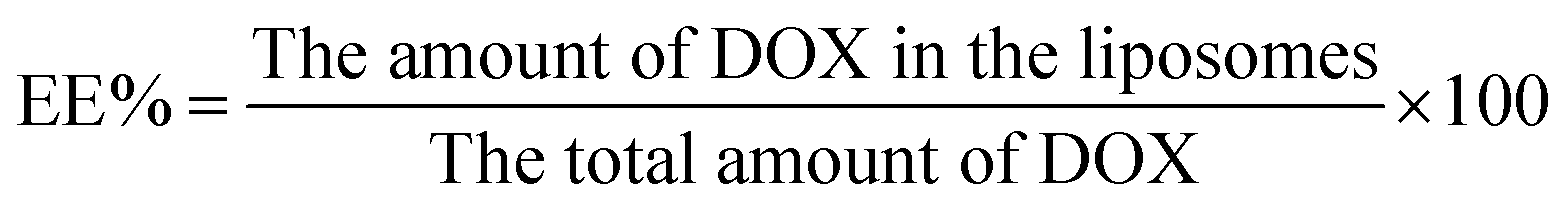
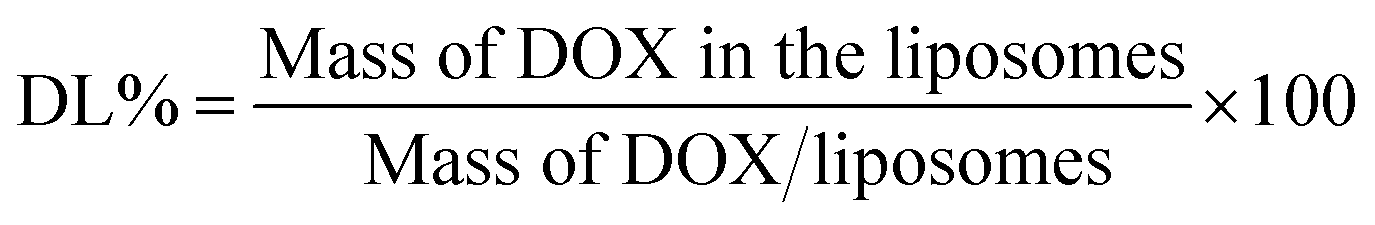
2.5. Drug release studies
In vitro release behaviour of DOX from liposomes was measured by a dialysis (MWCO 3.5 kDa) method. The release profiles of DOX from different liposomes were studied at 37 °C under pH 7.4 (normal pH) and pH 6.0 (tumor microenvironment pH). In brief, 2.0 ml of liposomes were added into the dialysis bag and placed it into a conical flask with 100 ml PBS solution in different pH. In order to meet sink condition, 0.5% Tween 80 was added into the release medium. All the flask were placed into a shaking incubator and the stirring speed was adjusted to 100 rpm. At different time point, 1.0 ml of the release medium was taken and equal volume of flash buffer was added to the flask. The amount of released DOX was measured using a UV spectrophotometer at 481 nm and cumulative amount of DOX release was calculated according to a standard calibration curve (data were not shown). All the release experiments were conducted in triplicate.2.6. In vitro cytotoxicity assay
2.7. Cellular uptake studies
2.8. Animal studies
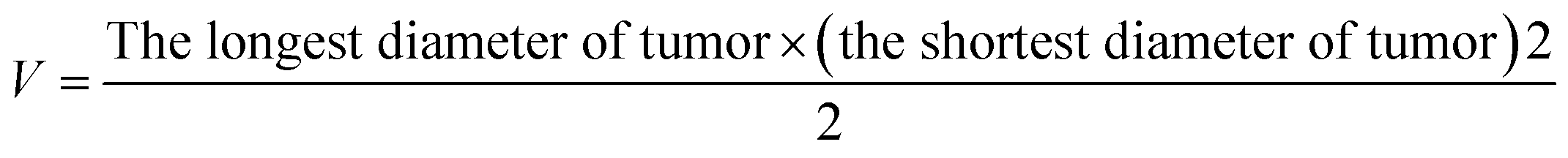
The antitumor activity was evaluated in terms of tumor weight (g). The tumor growth inhibition rate (IR) was calculated according to eqn:
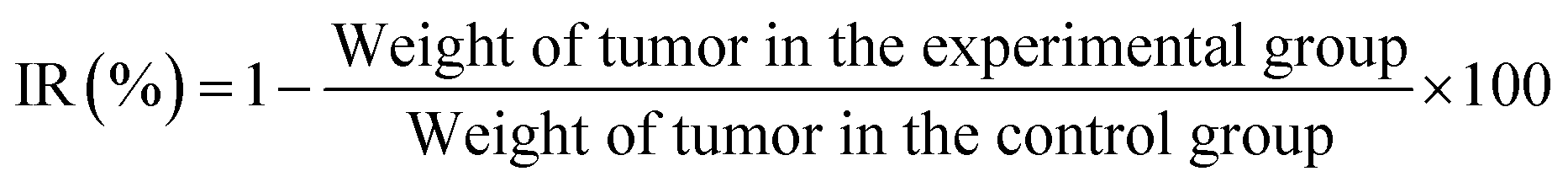
At day of 17, half of the mice were sacrificed and the tumor were harvested and other six mice were used to measure the survival curves. The survival time of all mice were recorded each day until all mice' death.
2.9. Statistical analysis
All experiments were performed at least three times. Quantitative data are presented as the mean ± SD. Statistical comparisons were determined by the analysis of variance (ANOVA) among at least three groups or Student's t-test between two groups. P-values < 0.05 and P-values < 0.01 were considered statistically significant.3 Results and discussion
3.1. Synthesis and characteristic of GPSC
To prepare the GA modified PEG cholesterol derivate (GPSC), we first synthesized Chol–SIB–NH2 by two step reactions as shown in Fig. 1. The first step consisted of introducing para-hydroxy benzaldehyde to the cholesterol molecule (Chol–CHO). In the second step, –SIB– group was synthesized by conjugating para-phenylenediamine to the terminal group of Chol–CHO (Chol–SIB–NH2). Multifunctional PEG (NH2–PEG2000–COOH) was used to modify the Chol–SIB–NH2 via amidation reaction. The –NH2 group was permitted to react with GA via amidation reaction in the end. As shown in Fig. 2, the formation of GPSC was confirmed by the 1H NMR spectrum and chemical shift values were described as follows: peaks of 1.07–2.03 ppm corresponding to the protons of cholesterol. The peaks at 0.7–1.59 ppm corresponding to the protons of GA unit. The chemical shift of PEG2000 was 3.70 ppm and the chemical shift of Schiff base bond of the pH-sensitive GPSC was 8.50 ppm. All these data confirmed that the novel pH-sensitive biomaterial GPSC was synthesized successfully.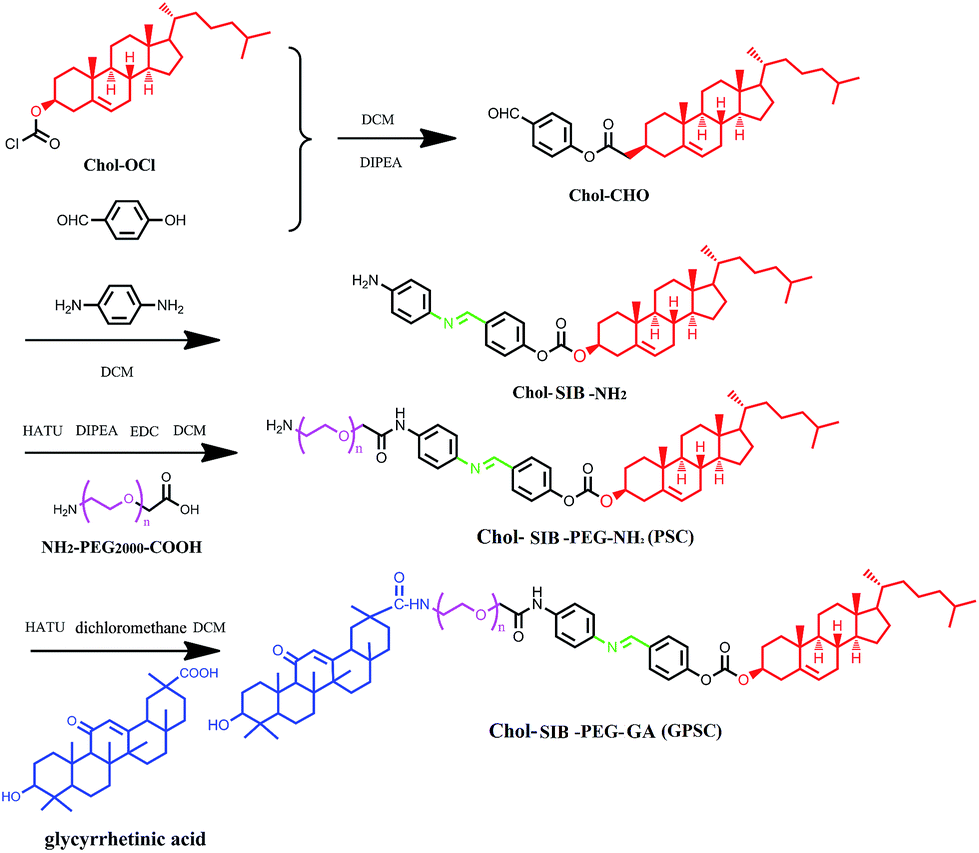 | ||
| Fig. 1 Synthesis pathway of GPSC. | ||
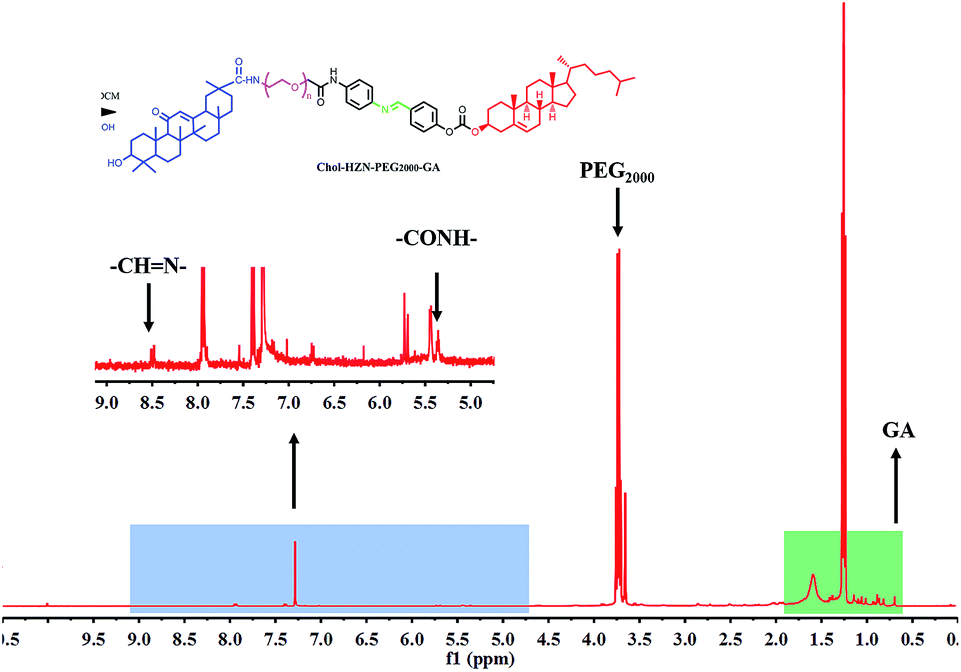 | ||
| Fig. 2 Typical 1H-NMR spectrum of GPSC in CDCl3. | ||
3.2. Characterization of liposomes
In this section, particle size, zeta potential, encapsulation efficiency (EE%) of different types of liposomes were analysed and presented in Table 1. There was no significant difference among mean encapsulation efficiency of different liposomes within the range of 53.8–58.5%. Although the EE% of PLP/DOX tended to be higher than GA–PEG–SIB–DOX liposomes, the difference was not statistically significant (P > 0.05). Compared with LP/DOX, there was a narrow increase in the mean particle size, which were 120.9 nm and 135.5 nm for PLP/DOX and GPSLP/DOX, respectively. In fact, zeta potential influences the liposomes stability in dispersion through electrostatic repulsion among particles. Compared with blank Lip and LP/DOX, there was a decrease in zeta potential of PLP/DOX and GPSLP/DOX, which were −4.59 mV and −5.19 mV, respectively. This interesting result was attributed to the charge shielding of PEG and GA outside on the surface of liposomes.43,44 Furthermore, the neutral or anionic surface charge of nanoparticles could efficiently escape from the renal elimination.45 Reticuloendothelial system (RES) could destroy any foreign particles through opsonisation. Hence, the liposomes must be hidden from the RES to circulate for a sufficient time in vivo, which could reach accumulation in tumor sites. Therefore, in this study, the liposomes with anionic charge could efficiently escape from RES and accumulated to the tumor.| Particle size (nm) | Polydispersity index | Zeta potential (mV) | EE% | LC% | |
|---|---|---|---|---|---|
| Blank Lip | 101.5 ± 2.3 | 0.103 ± 0.03 | −10.23 ± 0.68 | — | |
| LP/DOX | 110.2 ± 4.5 | 0.212 ± 0.06 | −7.45 ± 1.12 | 58.45 ± 1.5 | 2.62 ± 0.31 |
| PLP/DOX | 120.9 ± 1.8 | 0.254 ± 0.04 | −4.59 ± 0.39 | 56.41 ± 2.3 | 2.51 ± 0.24 |
| GPSLP/DOX | 135.5 ± 2.6 | 0.243 ± 0.08 | −5.19 ± 0.73 | 53.83 ± 5.8 | 2.39 ± 0.26 |
The morphology of the liposomes were observed through transmission electron microscope (TEM) as shown in Fig. 3. Results showed that the liposomes were homogeneously distributed spheroids with narrow size distribution. The particle size determined from transmission electron microscope was in line with the size measured by dynamic light scattering (Table 1). The particle size plays a vital role in in vivo circulation and tissue targeting based on receptor-mediated endocytosis.
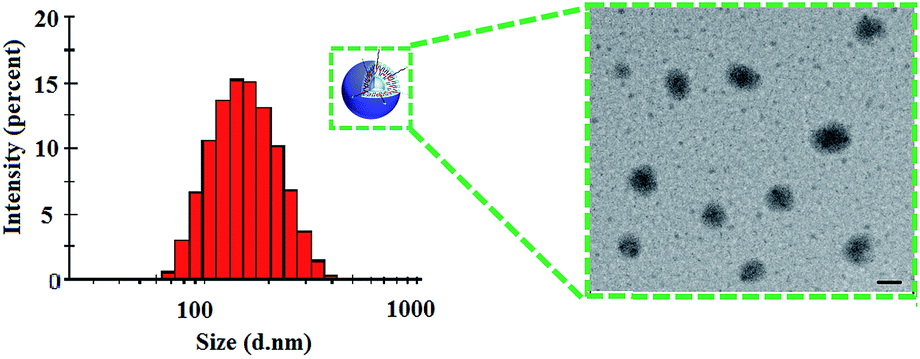 | ||
| Fig. 3 Size distribution and transmission electron microscopy of GPSLP/DOX. Scale bar represents 100 nm. | ||
3.3. In vitro release assay
As we all known, after reaching to target site, the nano-vectors would face in an acidic condition (pH value range from 6.8 to 5.5) during the process of endocytosis.46,47 In order to improve the endocytosis efficiency of the GPSLP/DOX, PEG was conjugated to the cholesterol by Schiff base bonds, which showed pH-responsive in such weak acidic environment. In vitro drug release profiles of DOX were studied using dialysis method in pH 7.4 (blood circulation pH) and pH 6.0 (tumor microenvironment pH). The in vitro accumulative release percentage of DOX from DOX solution and liposomes were shown in Fig. 4, DOX solution rapidly released up to nearly 100% in 10 hours in PBS (pH7.4). In addition, there was no significant difference between the release profiles of PLP/DOX in pH 7.4 and pH 6.0 with only about 50% of the accumulation release due to the presence of PEG on the outer surface of liposomes, which might hinder the release of DOX. This phenomenon also clarified the importance of PEG chains in the surface stability of liposomes. For the GPSLP/DOX, it could be observed that the drug release noticeably increased about 20% more in pH 6.0 than pH 7.4 over 72 h, indicating significantly pH-sensitivity of the designed liposome. There was no significant difference between PLP/DOX and GPSLP/DOX in pH 7.4. However, the accumulative release percentage of GPSLP/DOX was higher than PLP/DOX in pH 6.0, which showed significantly pH-sensitive feature. Overall, the release rate of DOX from GPSLP/DOX increased as the pH decreased, demonstrating that the introduction of GPSC on liposomes greatly affected the release behaviour of DOX from liposomes. The probable explanation for this phenomenon was that long PEG chains inserted on the surface of liposomes hindered the drug releasing from liposomes in pH 7.4, whereas, PEG gradually deviated from the liposome surface in pH 6.0 (slightly acidic environment) and the amount of released drugs increased as well. These results suggested that this liposomal delivery system could retain DOX while circulating in blood plasma and normal liver tissue, thereby reducing the systematic toxicity. This is expected to limit the leakage of DOX in plasma circulation and relieve the nonspecific adverse effects caused by DOX. Moreover, GPSLP/DOX had a superior release profile at pH 6.0 compared to conventional PEG long circulation liposomes (PLP/DOX), indicating that DOX could release in the acidic tumor microenvironment or endosomes in cytoplasm. This phenomenon would ensure effective intracellular delivery and lead to a high concentration of DOX in the tumor cells, which resulted in a higher cytotoxicity to the tumour cells. Thus the enhanced amount of drug release from liposomes might improve the therapeutic potential of DOX in tumor cells.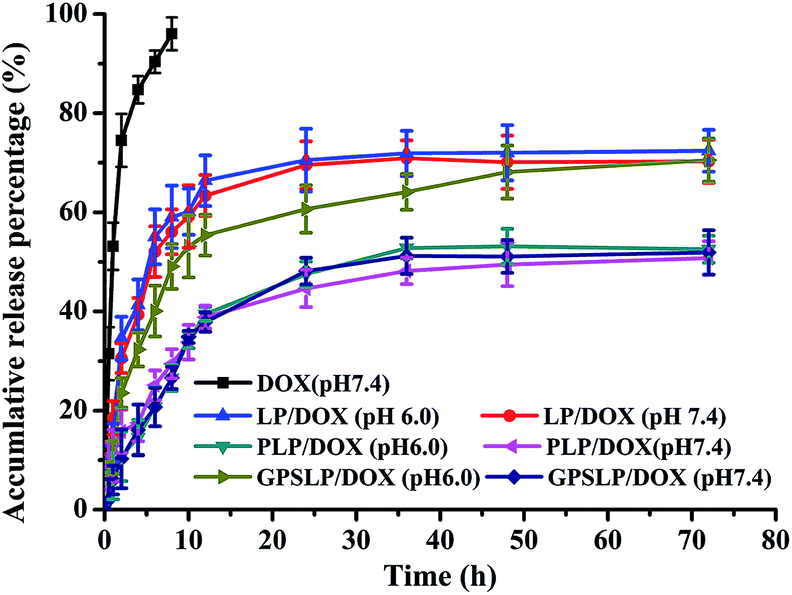 | ||
| Fig. 4 pH-sensitive release of DOX from different formulations at 37 °C (n = 3). | ||
3.4. In vitro cytotoxicity assay
The cytotoxicity of free DOX solution, PLP/DOX, GPSLP/DOX and corresponding blank liposomes was investigated in HepG2 cells using standard MTT method. The viability of cells was measured immediately after stopping incubation. As revealed in Fig. 5a, with an increase in concentration, there was no significant cytotoxicity observed for blank liposomes against HepG2 cells at the concentrations of 100–1000 μg ml−1, reflecting that blank liposomes were biocompatible and suitable for a potential drug delivery system.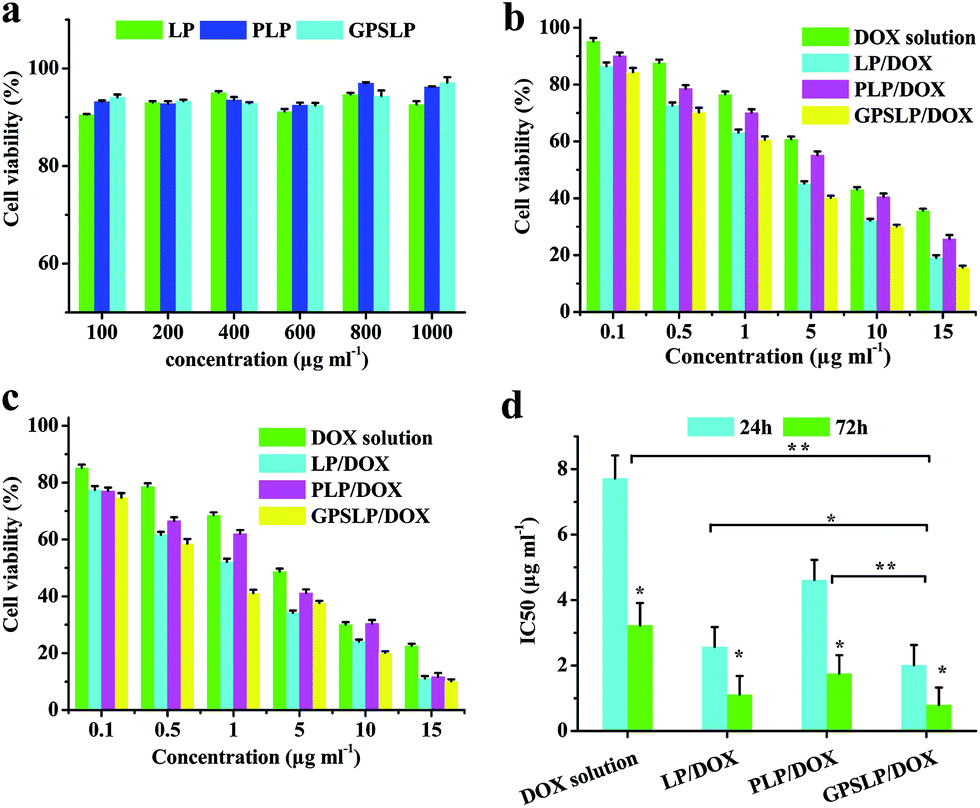 | ||
| Fig. 5 (a) In vitro cytotoxicity of blank liposomes treating with HepG2 cells; (b and c) MTT assay of different formulation in 24 and 72 h, respectively, against HepG2 cells (n = 3); (d) IC50 of different formulation in 24 and 72 h. *p < 0.05, **p < 0.01. | ||
For the MTT studies of different DOX loaded liposomes, cell viability decreased as the dose increased, which showed significant dose-independent feature. The cell cytotoxicity of different formulation has been showed in the Fig. 5b and c. From the cytotoxicity data we could find that compared with DOX solution group, LP/DOX group showed higher cytotoxicity against cells no matter in 24 h or in 72 h. This phenomenon was probably because the hydrophobic lipid membrane of liposomes could enhance the cellular uptake to reach higher cytotoxicity. In addition, the cytotoxicity of PLP/DOX groups showed lower cytotoxicity compared with LP/DOX group. This was attributed to the hydrophilic shell of PEG would form a weak exclusion between cell membrane and formulation, which resulted in a lower cytotoxicity. Interestingly, there was significant difference between GPSLP/DOX group and other groups. As shown in Fig. 5d, the IC50 of GPSLP/DOX were 2.01 ± 0.61 and 0.80 ± 0.53 μg ml−1 for 24 h and 72 h, respectively. The higher cytotoxicity of GPSLP/DOX groups was probably two reasons: (1) GA mediated cell endocytosis could enhance the cellular uptake of the formulation. (2) Due to the pH-sensitive PEG shell could increase the drug release from the preparation and a large amount of DOX could efficiently release to the cytoplasm, which resulted in a higher therapeutical effect.
3.5. Cellular uptake assay
In this study, CLSM was used to evaluate the cellular uptake of different formulations. As shown in Fig. 6a, there was only a weak fluorescence signal in all groups when the incubation time was approximately 0.5 h. When the incubation time increased, red fluorescence intensity increased in different degrees. Compared with DOX solution group, there was higher fluorescence intensity among all other groups. The intracellular DOX was increased in the cytoplasm and a large amount of DOX was released from the liposomes and DOX was delivered to the cytoplasm. Among all the groups, GPSLP/DOX group showed the highest fluorescence intensity. GA receptor-mediated cell endocytosis and pH-responsive rapid drug release were probably the main reason to explain this result. This results indicated that the cellular uptake of different formulation was time-independent.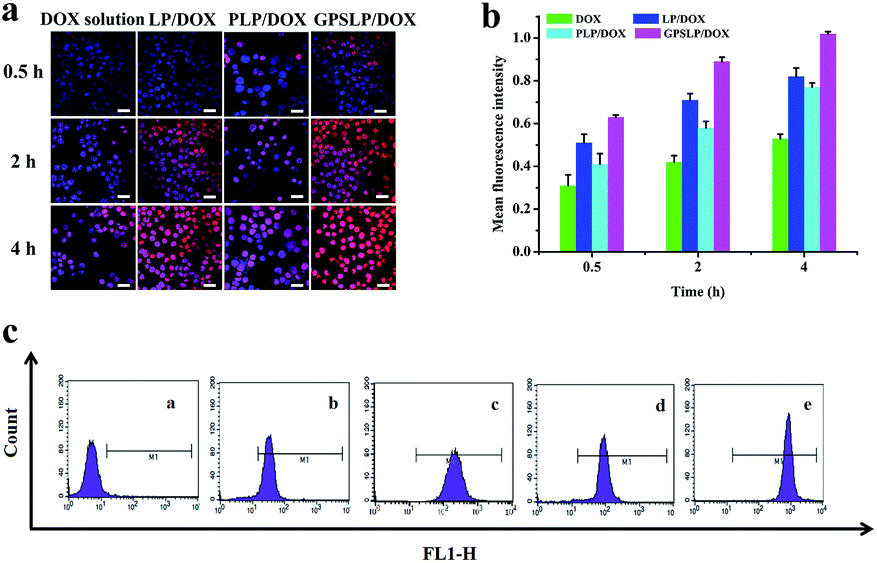 | ||
| Fig. 6 (a) Fluorescence microscopy images of HepG2 cells incubated with different formulations at different time point. Red and blue colors indicate DOX and Hoechst 33258, respectively. Scale bar represents 50 μm. (b) Flow cytometry measurement of cellular uptake of different formulation at different time point. (c) Fluorescence intensity of different formulation at 4 h. (a), (b), (c), (d), (e) were control, DOX solution, LP/DOX, PLP/DOX and GPSLP/DOX, respectively. | ||
Intracellular uptake efficiency of different liposomes was also quantitatively analysed by flow cytometry. As was illustrated in Fig. 6b, compared with free DOX group, the cellular uptake of different liposomes significantly increased in HepG2 cells when the incubation time was 4 h. There was significant difference between LP/DOX and PLP/DOX group in every time point, indicating that the PEG shell could decrease the cellular uptake in different degree. However, there was a significantly increase in fluorescence intensity for GPSLP/DOX group in every time point, which was 1.92-, 1.24- and 1.32-fold higher than DOX, LP/DOX, PLP/DOX group, respectively (Fig. 6c). This result was in consistent with the CLSM, indicating that the GA modified could dramatically increase the affinity between the liposomes and the cell membrane.
All the results of quantitative and qualitative analysis of cellular uptake demonstrated that the GPSLP/DOX liposomes had better cell selectivity and stronger internalization than non-targeted PLP/DOX. The GPSLP/DOX could specifically recognize cells through overexpressing targeting binding sites of GA receptors and mediate efficiently internalization in HepG2 cells.
3.6. In vivo biodistribution studies
In order to evaluate the in vivo biodistribution of different formulations in nude mice bearing H22 tumor cells. In vivo fluorescence imaging study was carried out in this section. DiR reagent was used to observe the biodistribution behaviour of different formulations. As shown in Fig. 7, during the first 12 h, the fluorescence signal of DiR solution group showed whole body biodistribution after administration through tail vein. However, the fluorescence intensity decreased dramatically and the intensity became weaker in liver and spleen at 24 h. There was only a weak fluorescence signal was observed in 72 h, indicating that most of DiR was eliminated through RES. In comparison, the biodistribution behaviour of different formulations were not the same as DiR solution group. There was strong fluorescence intensity in tumor and liver for both LP/DOX group and PLP/DOX group on the first 12 h due to the EPR effect. For LP/DOX group, the signal became weaker in both tumor and liver in 72 h, this phenomenon was probably because LP/DOX was recognized by RES and eliminated in liver or some other organs. Hopefully, PLP/DOX showed significant higher intensity in tumor site, this result confirmed that PEG shell could avoid the reorganization of RES to reach long circulation feature. As was expected, GPSLP/DOX showed the best targetability in tumor sites among all the groups. The exciting result was probably attributed to pH-sensitive PEG shell was stable in blood circulation, which resulted in a lower drug leakage rate and rapid release in acidic microenvironment. In addition, GA, modified on the surface of the liposomes played an important role in the target the tumor sits. These advantages of GPSLP/DOX attributed to the higher targetability in the in vivo biodistribution assay.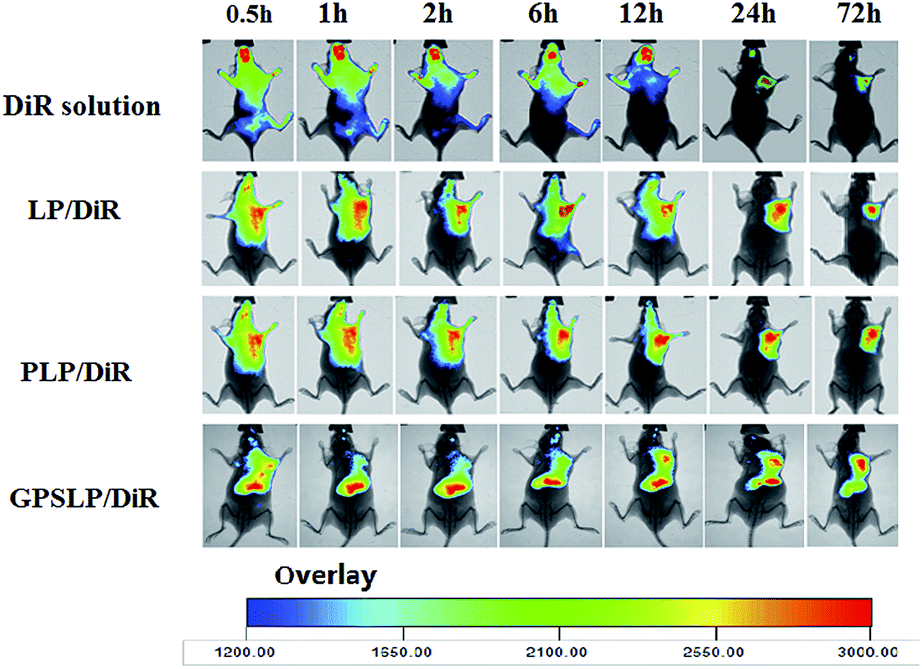 | ||
| Fig. 7 In vivo whole body imaging of H22 tumor-bearing nude mice after free DiR solution and DiR liposomes administration at different time point with the same dose of DiR (20 μg ml−1). | ||
3.7. In vivo antitumor activity
In vivo antitumor activities of the DOX-loaded liposomes were assessed in mice bearing H22 liver tumor at a dose of 5 mg kg−1. The average tumor volumes and changes of body weights were monitored during the experiment to observe the antitumor efficiency and toxicity, respectively. Fig. 8a showed the tumour growth inhibition of drug loaded liposomes. As expected, control group showed a rapid increase of tumor size within experimental period, whereas, DOX solution treated group just showed a slightly inhibition in tumour growth. Hopefully, all the DOX-loaded liposomal formulations suppressed the growth of tumours effectively. Particularly, GPSLP/DOX could inhibit the tumor growth more efficiently than other liposomal groups with the tumour growth inhibition rate (IR) reached to 81.76%. The higher tumor growth inhibition ratio of GPSLP/DOX was resulted from several reasons: (1) PEG shell modified on the surface of the liposomes could reach long circulation effect and avoid elimination by RES. (2) GA-mediated cell endocytosis could increase the accumulation of DOX in tumour tissues. (3) pH-responsive drug release from liposomes could reduce systematic toxicity and increase therapeutical effect. Therefore, we have reasons to suppose that the higher therapeutic potential is due to the high DOX concentration accumulated in tumours.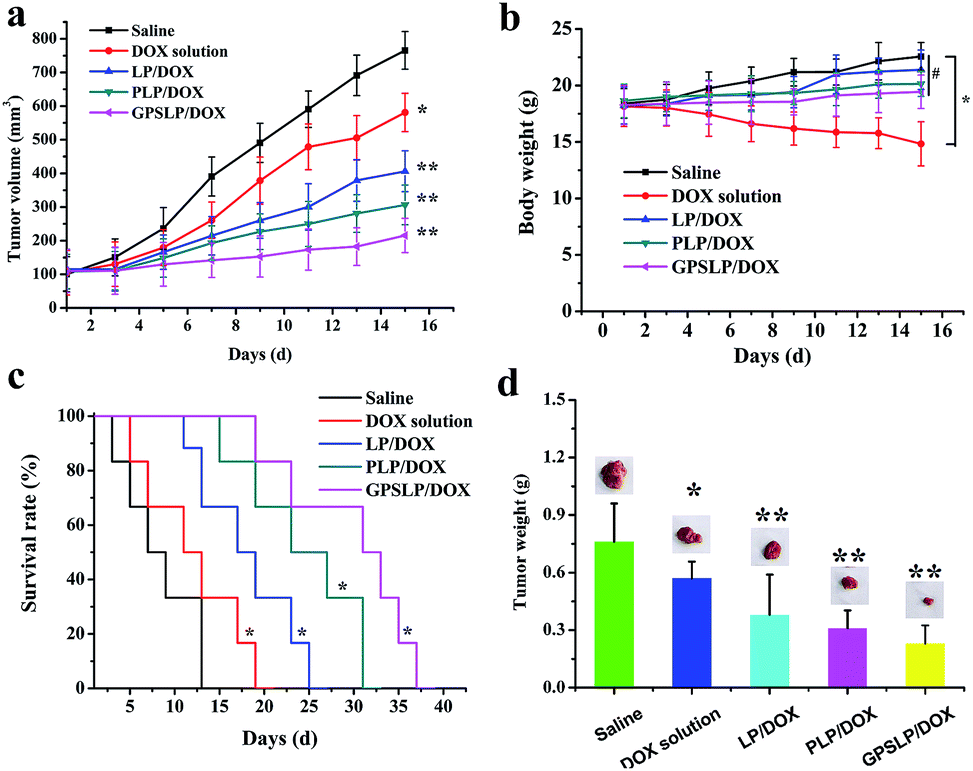 | ||
| Fig. 8 The mean tumor volume (a), body weight (b), Kaplan–Meier survival curve (c) and tumor inhibition rate (d) of different formulation in nude mice bearing H22 tumor cells on intravenous administration of different formulation (n = 6). *p < 0.05, **p < 0.01, # not significant. | ||
Fig. 8b described the change of body weight after treated with saline, DOX solution and three different liposomal groups. Body weight of liposomal formulation groups and saline group showed a slightly increase during treatment and no significant difference was observed with each other (P > 0.05). In contrast, significant weight loss (P < 0.001) was observed in DOX solution treated mice compared to control and the liposomal groups. This result was probably free DOX showed whole-body biodistribution behaviour, which led to systematic toxicity. In contrast, sustained release of DOX from liposomes showed less systemic toxicity as evidenced by no loss of body weight among three liposomal groups. Half of mice were sacrificed and tumours were taken out after 15 days of treatment. Fig. 8d showed the tumour weight of each group on the 15th day. The tumor size of control group was the biggest, and the smaller was free DOX group, followed by LP/DOX group and PLP/DOX group. In contrast, the tumor size of GPSLP/DOX group was the smallest.
At the end of the experiment, Kaplan–Meier survival curve was chosen as a method to verify the in vivo antitumor activities of different formulation. As shown in Fig. 8c, after administration of different formulation for several times, the survival time of control group was 15 days. Compared with control group, DOX solution showed a slightly higher survival time, which was 21 days. This was probably because although DOX could inhibit tumor growth, the nonspecific toxicity would also decrease its survival time. In comparison, three different formulation groups showed a significantly higher survival time, which was 27 days, 33 days and 39 days for LP/DOX, PLP/DOX and GPSLP/DOX groups, respectively. Among all of them, GPSLP/DOX group showed the highest survival time, this result was because the active target molecule GA modified on the surface of the liposomes could increase the affinity between liposomes and the tumor sites, thus decreased the systematic toxicity. In addition, pH-responsive rapid drug release in tumor microenvironment could also increase the therapeutical effect and increase its survival time.
Based on all the in vitro and in vivo studies, it was confirmed that the active liver targeted pH sensitive liposomes (GPSLP/DOX) could successfully elicit the desired pharmacological and therapeutic responsive without acute adverse effect.
4 Conclusions
In this study, a novel pH-sensitive conjugate GPSC was successfully synthesized and applied as active targeted composition to establish the GPSLP/DOX for targeting therapy of liver cancer. These liposomes exhibited promise in delivering therapeutic doses of DOX to hepatocellular carcinoma cells expressing GA receptors. In vivo antitumor study showed that the systematic toxicity of DOX was reduced for all the DOX-loaded liposomes. Furthermore, GPSLP/DOX showed the highest tumor suppression. In conclusion, GPSLP/DOX are promising drug carriers for liver cancer therapy.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (81202483) as well as the program for Liaoning Natural Science Foundation (LR 2015020543), Scientific Research Foundation for the Rturuned Overseas Chinese Scholars by State Education Ministry (201303003), Science and Technology project of Shenyang (F15-199-1-24, F15-139-9-06).References
- F. Islami, R. Dikshit, M. K. Mallath and A. Jemal, Cancer Epidemiol., 2016, 40, 79–86 CrossRef PubMed
.
- M. Sassier, A. E. Dugué, B. Clarisse, P. Lesueur, V. Avrillon, A. Bizieux-Thaminy, J.-B. Auliac, L. Kaluzinski, J. Tillon, G. Robinet, H. Le Caer, I. Monnet, A. Madroszyk, G. Boza, L. Falchero, P. Fournel, T. Egenod, A.-C. Toffart, N. Leiber, P. Do and R. Gervais, Lung Cancer, 2015, 89, 161–166 CrossRef PubMed
- P. Strati, L. V. Abruzzo, W. G. Wierda, S. O'Brien, A. Ferrajoli and M. J. Keating, Clin. Lymphoma, Myeloma Leuk., 2015, 15, 420–427 CrossRef PubMed
- L. Tong, C. Ahn, E. Symanski, D. Lai and X. L. Du, Ann. Epidemiol., 2014, 24, 411–417 CrossRef PubMed
- K. Beaver, S. Williamson and J. Briggs, Eur. J. Oncol. Nurs., 2016, 20, 77–86 CrossRef PubMed
- A. Charehbili, N. A. T. Hamdy, V. T. H. B. M. Smit, L. Kessels, A. van Bochove, H. W. van Laarhoven, H. Putter, E. Meershoek-Klein Kranenbarg, A. E. van Leeuwen-Stok, J. J. M. van der Hoeven, C. J. H. van de Velde, J. W. R. Nortier and J. R. Kroep, Breast, 2016, 25, 69–74 CrossRef CAS PubMed
- L. B. Piacentine, J. F. Miller, S. Haberlein and A. S. Bloom, Appl. Nurs. Res., 2016, 29, 9–11 CrossRef PubMed
- F. Khemissa, L. Mineur, C. Amsellem, E. Assenat, M. Ramdani, P. Bachmann, C. Janiszewski, I. Cristiani, F. Collin, J. Courraud, H. de Forges, P. Dechelotte and P. Senesse, Dig. Liver Dis., 2015 DOI:10.1016/j.dld.2015.11.025
- B. Sjøblom, B. H. Grønberg, J. Š. Benth, V. E. Baracos, Ø. Fløtten, M. J. Hjermstad, N. Aass and M. Jordhøy, Lung Cancer, 2015, 90, 85–91 CrossRef PubMed
- W.-H. Chen, G.-F. Luo, Q. Lei, F.-Y. Cao, J.-X. Fan, W.-X. Qiu, H.-Z. Jia, S. Hong, F. Fang, X. Zeng, R.-X. Zhuo and X.-Z. Zhang, Biomaterials, 2016, 76, 87–101 CrossRef CAS PubMed
- J. Depreeuw, E. Hermans, S. Schrauwen, D. Annibali, L. Coenegrachts, D. Thomas, M. Luyckx, I. Gutierrez-Roelens, D. Debruyne, K. Konings, P. Moerman, I. Vergote, D. Lambrechts and F. Amant, Gynecol. Oncol., 2015, 139, 118–126 CrossRef PubMed
- F. Zhang, X.-Q. Kong, Q. Li, T.-T. Sun, C. Chai, W. Shen, Z.-Y. Hong, X.-W. He, W.-Y. Li and Y.-K. Zhang, Talanta, 2016, 148, 108–115 CrossRef CAS PubMed
- G. A. T. Kaminski, M. R. Sierakowski, R. Pontarolo, L. A. d. Santos and R. A. d. Freitas, Carbohydr. Polym., 2016, 140, 129–135 CrossRef CAS
- Y. Mao, X. Li, G. Chen and S. Wang, J. Pharm. Sci., 2016, 105, 194–204 CrossRef PubMed
- C. Tan, B. Feng, X. Zhang, W. Xia and S. Xia, Food Hydrocolloids, 2016, 52, 774–784 CrossRef CAS
- M. W. Jøraholmen, N. Škalko-Basnet, G. Acharya and P. Basnet, Eur. J. Pharm. Sci., 2015, 79, 112–121 CrossRef PubMed
- H. Shibata, H. Yoshida, K.-I. Izutsu, Y. Haishima, T. Kawanishi, H. Okuda and Y. Goda, Int. J. Pharm., 2015, 495, 827–839 CrossRef CAS PubMed
- S. Bersani, M. Vila-Caballer, C. Brazzale, M. Barattin and S. Salmaso, Eur. J. Pharm. Biopharm., 2014, 88, 670–682 CrossRef CAS PubMed
- H. Li and F. Chen, Saudi J. Biol. Sci., 2015 DOI:10.1016/j.sjbs.2015.10.025
- Y. Chen, L. V. Minh, J. Liu, B. Angelov, M. Drechsler, V. M. Garamus, R. Willumeit-Römer and A. Zou, Colloids Surf., B, 2016, 140, 74–82 CrossRef CAS PubMed
- H. S. Oberoi, Y. M. Yorgensen, A. Morasse, J. T. Evans and D. J. Burkhart, J. Controlled Release, 2016, 223, 64–74 CrossRef CAS PubMed
- E. Marzban, S. H. Alavizadeh, M. Ghiadi, M. Khoshangosht, Z. Khashayarmanesh, A. Abbasi and M. R. Jaafari, Colloids Surf., B, 2015, 136, 885–891 CrossRef CAS PubMed
- Y.-L. Lo and W.-C. Tu, Chem.-Biol. Interact., 2015, 242, 13–23 CrossRef CAS PubMed
- Y. Wan, J. Han, G. Fan, Z. Zhang, T. Gong and X. Sun, Biomaterials, 2013, 34, 3020–3030 CrossRef CAS PubMed
- Z. Zou, X. He, D. He, K. Wang, Z. Qing, X. Yang, L. Wen, J. Xiong, L. Li and L. Cai, Biomaterials, 2015, 58, 35–45 CrossRef CAS PubMed
- D. Kokuryo, S. Nakashima, F. Ozaki, E. Yuba, K.-H. Chuang, S. Aoshima, Y. Ishizaka, T. Saga, K. Kono and I. Aoki, Nanomedicine, 2015, 11, 229–238 CrossRef CAS PubMed
- M. Yu, F. Guo, F. Tan and N. Li, J. Controlled Release, 2015, 215, 91–100 CrossRef CAS PubMed
- H. Fu, K. Shi, G. Hu, Y. Yang, Q. Kuang, L. Lu, L. Zhang, W. Chen, M. Dong, Y. Chen and Q. He, J. Pharm. Sci., 2015, 104, 1160–1173 CrossRef CAS PubMed
- J.-W. Choi, S.-J. Jung, D. Kasala, J. K. Hwang, J. Hu, Y. H. Bae and C.-O. Yun, J. Controlled Release, 2015, 205, 134–143 CrossRef CAS PubMed
- J. Song, Z. Ge, X. Yang, Q. Luo, C. Wang, H. You, T. Ge, Y. Deng, H. Lin, Y. Cui, W. Chu, M. Yao, Z. Zhang, J. Gu, J. Fan and W. Qin, Cancer Lett., 2015, 356, 713–720 CrossRef CAS PubMed
- L. Qiu, Z. Li, M. Qiao, M. Long, M. Wang, X. Zhang, C. Tian and D. Chen, Acta Biomater., 2014, 10, 2024–2035 CrossRef CAS PubMed
- P. Ghosh, N. Kumar, S. K. Mukhopadhyay and P. Banerjee, Sens. Actuators, B, 2016, 224, 899–906 CrossRef CAS
- A. A. Abdel Aziz, S. H. Seda and S. F. Mohammed, Sens. Actuators, B, 2016, 223, 566–575 CrossRef CAS
- L. Zhang, J.-P. Zhou and J. Yao, Chin. J. Nat. Med., 2015, 13, 915–924 Search PubMed
- S.-Z. Kong, H.-M. Chen, X.-T. Yu, X. Zhang, X.-X. Feng, X.-H. Kang, W.-J. Li, N. Huang, H. Luo and Z.-R. Su, Exp. Gerontol., 2015, 61, 147–155 CrossRef CAS PubMed
- H. Du, M. Liu, X. Yang and G. Zhai, J. Colloid Interface Sci., 2015, 460, 87–96 CrossRef CAS PubMed
- C. Zhang, W. Wang, T. Liu, Y. Wu, H. Guo, P. Wang, Q. Tian, Y. Wang and Z. Yuan, Biomaterials, 2012, 33, 2187–2196 CrossRef CAS PubMed
- W. Huang, W. Wang, P. Wang, Q. Tian, C. Zhang, C. Wang, Z. Yuan, M. Liu, H. Wan and H. Tang, Acta Biomater., 2010, 6, 3927–3935 CrossRef CAS PubMed
- F. Versluis, J. Voskuhl, B. van Kolck, H. Zope, M. Bremmer, T. Albregtse and A. Kros, J. Am. Chem. Soc., 2013, 135, 8057–8062 CrossRef CAS PubMed
- H.-Y. Wang, H.-R. Jia, X. Lu, B. Chen, G. Zhou, N. He, Z. Chen and F.-G. Wu, J. Mater. Chem. B, 2015, 3, 6165–6173 RSC
- H.-Y. Wang, X.-W. Hua, H.-R. Jia, P. Liu, N. Gu, Z. Chen and F.-G. Wu, J. Mater. Chem. B, 2016, 4, 834–843 RSC
- Y. Teramura, K. Kuroyama and M. Takai, Acta Biomater., 2016, 30, 135–143 CrossRef CAS PubMed
- H. Tian, Z. Guo, L. Lin, Z. Jiao, J. Chen, S. Gao, X. Zhu and X. Chen, J. Controlled Release, 2014, 174, 117–125 CrossRef CAS PubMed
- B.-H. Im, J.-H. Jeong, M. R. Haque, D. Y. Lee, C.-H. Ahn, J. E. Kim and Y. Byun, Biomaterials, 2013, 34, 2098–2106 CrossRef CAS PubMed
- D. H. Jo, J. H. Kim, T. G. Lee and J. H. Kim, Nanomedicine, 2015, 11, 1603–1611 CrossRef CAS PubMed
- L. Maldonado-Báez, C. Williamson and J. G. Donaldson, Exp. Cell Res., 2013, 319, 2759–2769 CrossRef PubMed
- V. Iacobucci, F. Di Giuseppe, T. T. Bui, L. S. Vermeer, J. Patel, D. Scherman, A. Kichler, A. F. Drake and A. J. Mason, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1332–1341 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2016 |