
Intramolecular functional group differentiation as a strategy for the synthesis of bridged bicyclic β-amino acids†
Andriy V. Tymtsunikab,
Serhii O. Kokhanab,
Yevhen M. Ivonab,
Igor V. Komarovab and
Oleksandr O. Grygorenko*a
aNational Taras Shevchenko University of Kyiv, Volodymyrska Street 64, Kyiv 01601, Ukraine. E-mail: gregor@univ.kiev.ua
bEnamine Ltd., Alexandra Matrosova Street 23, Kyiv 01103, Ukraine
First published on 22nd February 2016
Abstract
Differentiation of identical electrophilic functional groups (carboxylates) by a strategically placed internal nucleophile (an amino group) in cyclic precursors was used as a key general approach to functionalized azabicyclic scaffolds. The utility of the method was demonstrated by the synthesis of three bicyclic β-amino acids (analogues of nipecotic acid), which were prepared in good yields and on a relatively large scale.
Introduction
Saturated bicyclic molecular scaffolds attract the attention of organic chemists not only because of their practical significance, but also as challenging synthetic targets that are among the structures associated with “chemical beauty”.1 Bicyclic diamines,2 α-3 and β-amino acids4 are examples of the targets that have found various applications in total syntheses of natural compounds, medicinal chemistry and the design of peptidomimetics. In particular, bicyclic β-amino acids, known since XIX century as constituents of tropane alkaloids (e.g. cocaine (1)), were used in total syntheses of alkaloids epibatidine (2), anatoxin A (3), and ibogamine (4). Numerous synthetic biologically active compounds were also prepared from bicyclic β-amino acids for example, α7 nAChR agonist 5, potent selective melanocortin subtype-4 receptor (MC4R) agonist RY764 (6), antagonist of inhibitors of apoptosis proteins cIAP1 (7), several PET/SPECT tracers (e.g. [18F]FE@CIT, 8) used for in vitro and in vivo visualization of dopamine transporter (DAT), or ligand of norepinephrine transporter (NET) 9 (Fig. 1).4b | ||
| Fig. 1 Biologically active compounds prepared from bicyclic β-amino acids (the fragments originated from them are highlighted in red). | ||
In general, construction of any bicyclic bifunctional scaffold is a synthetic task which is usually solved by developing specific retrosynthetic approach. Approaches addressing the challenge of preparation of bi- and polycyclic molecules by a general diversity-oriented synthesis (DOS) strategy were also reported.5 In a series of recent papers, we have described tandem Strecker reaction – nucleophilic cyclization (STRINC) sequence as a general approach to bicyclic α-amino acids.6 In this work, we report a different strategy where a cyclic trifunctional building block undergoes cyclization via intramolecular nucleophilic attack at one of two identical functional groups present in the molecule, leading to differentiation of the groups and formation of bicyclic scaffolds (Scheme 1).
 | ||
| Scheme 1 The key step of intramolecular functional group differentiation strategy. | ||
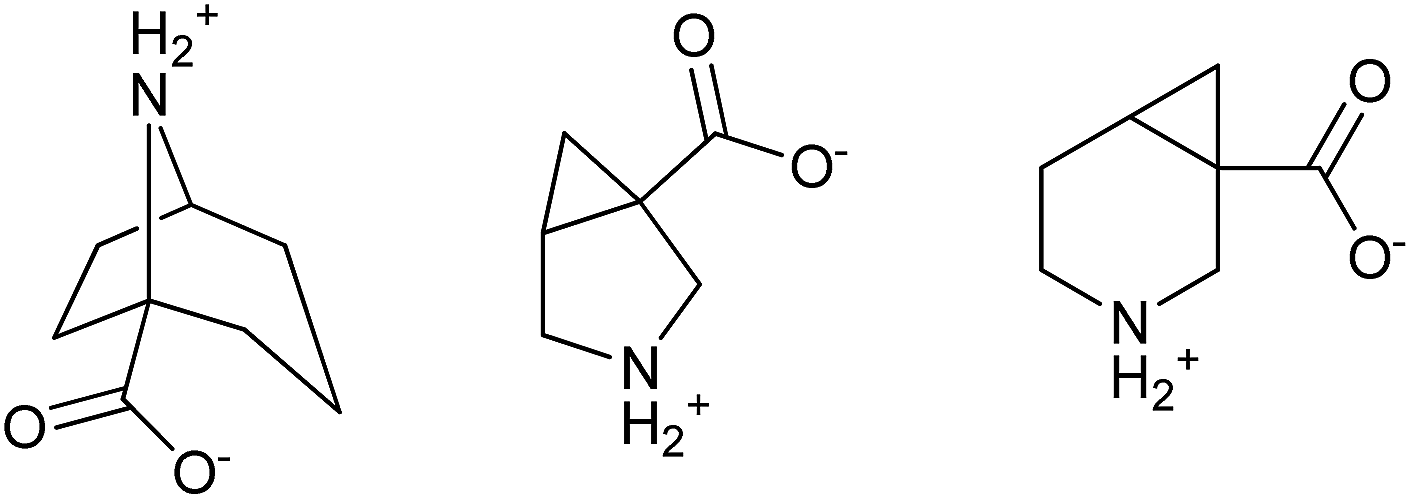
The main beneficial feature of such strategy is avoiding the problems related to formation and separation of stereoisomeric mixtures, as the internal nucleophile is at the position enabling reaction with only one electrophilic group. It should be noted that examples of using this approach for synthesis of bicyclic scaffolds can be found in the literature (Fig. 2).7 Here we demonstrate its generality by preparation of three novel β-amino acids 10–12, which are bridged bicyclic analogues of nipecotic acid (13) (Fig. 3).8
 | ||
| Fig. 2 Bicyclic α- and β-amino acids prepared using intramolecular functional group differentiation strategy (literature data). | ||
 | ||
| Fig. 3 Nipecotic acid (13) and its bridged bicyclic analogues 10–12. | ||
Results and discussion
Retrosynthetic analysis of the amino acids 10–12 according to the intramolecular functional group differentiation strategy leads to corresponding amino esters of the type 14–16 (Scheme 2) as the key intermediates. Further retrosynthetic transformations of 14–16 lead to the known functionalized diesters 17–19. | ||
| Scheme 2 Retrosynthetic analysis of the amino acids 10–12. | ||
Compound 17 was obtained by a known procedure in 53% overall yield starting from dibromide 20, using alkylation of diisopropyl malonate for the construction of the cyclobutane ring (Scheme 3).9 For preparation of the hydroxy diesters 18 and 19, we have developed our own methods which were also based on classical malonate chemistry. Compound 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10,11 was prepared from the known diol 21, in turn obtained in three steps from dimethyl acetonedicarboxylate (22) (Scheme 4).12 Compound 21 was transformed into dimesylate 23, which was used for the alkylation of dimethyl malonate to give the diester 24. The target hydroxy diester 18 was obtained from 24 upon fluoride-promoted deprotection.
10,11 was prepared from the known diol 21, in turn obtained in three steps from dimethyl acetonedicarboxylate (22) (Scheme 4).12 Compound 21 was transformed into dimesylate 23, which was used for the alkylation of dimethyl malonate to give the diester 24. The target hydroxy diester 18 was obtained from 24 upon fluoride-promoted deprotection.
 | ||
| Scheme 3 Synthesis of the intermediate 17. | ||
 | ||
| Scheme 4 Synthesis of the intermediate 18. | ||
Synthesis of the diester 19 aimed at the non-racemic product, as we wanted to synthesize the target amino acid 12 (the only chiral compound of 10–12) in optically pure form. The known methods for preparation of 19 lead to racemate;13 enantiopure 19 was obtained before our work only as a minor product.14 Our approach to both enantiomers of 19 was similar to that described by Ma and co-workers for the corresponding (S)-ethyl ester;15 in our version, we focused on developing a practical scaled-up procedure. The synthesis commenced from natural (S)-malic acid (25), which was transformed into dimethyl malate (26) in nearly quantitative yield by refluxing with strong cation exchange resin in methanol (Scheme 5).
 | ||
| Scheme 5 Synthesis of diester (S)-19. | ||
Benzyl group was used for the protection of the hydroxyl moiety in 26, since the diphenylmethyl substituent described in the previous work was found unsuitable for scale-up. Alkylation of 26 with benzyl bromide was performed according to the described procedure using Ag2O in EtOAc, allowing preparation of the product 27 in 94% yield.16 Notably, silver-containing side products could be recovered in more than 90% yield after the synthesis, which is important for large-scale preparation. Further transformations included reducing of 27 with LiAlH4, which gave the known diol 28 (90%).17 Initially, we performed transformation of 28 into the corresponding cyclic sulfate; however, it turned out to be not reactive towards dimethyl malonate. Therefore, more reactive diiodide 29 was prepared in 91% yield by the method described previously.16
The final steps in the synthesis of key intermediate (S)-19 included the reaction of 29 with dimethyl malonate. This step required tedious optimization of the reaction conditions; it was found that the product 30 was formed in 80% yield if 2.5–3.5 excess of the nucleophile in refluxing THF was used. The product 30 could not be separated from the excess of dimethyl malonate preparatively neither by column chromatography nor by distillation (although we managed to obtain its analytical sample). Since dimethyl malonate did not interfere with the next step (catalytic debenzylation), the crude 30 was used without purification. After hydrogenation and subsequent column chromatography, (S)-enantiomer of the target product (S)-19 was obtained in 74% yield. The other enantiomer, (R)-19, was obtained from (S)-19 in two steps in 67% overall yield via Mitsunobu inversion (Scheme 6).
 | ||
| Scheme 6 Synthesis of the intermediate (R)-19. | ||
Transformation of both 18 and 19 into the amino diesters 15 and 16 was achieved by mesylation, nucleophilic substitution with NaN3, and catalytic hydrogenation (Scheme 7). Compound 15 was not isolated in pure form but used in the next step without purification; both enantiomers of 16 were prepared as hydrochlorides.
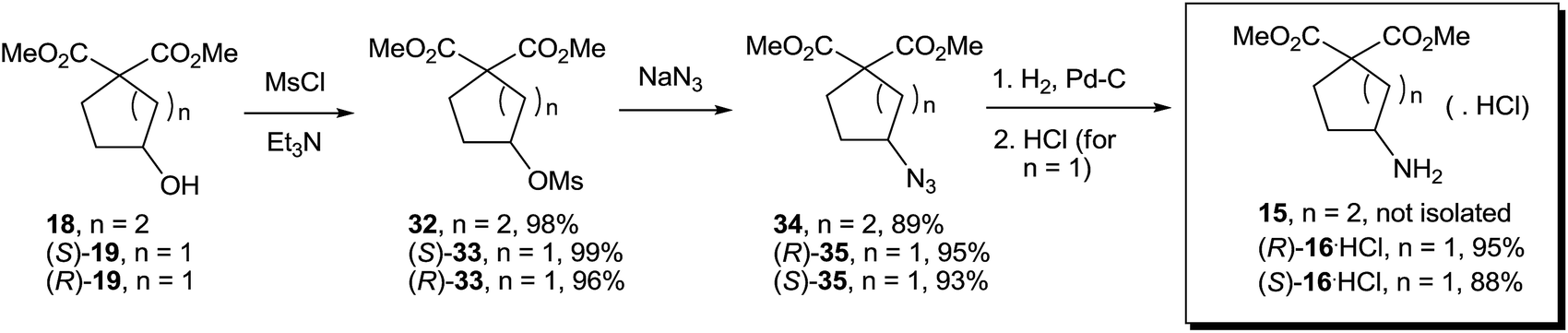 | ||
| Scheme 7 Synthesis of 15 and 16·HCl. | ||
To obtain the amino diester 14, elongation of the carbon chain in the molecule of 17 was done via Horner–Wadsworth–Emmons olefination, leading to the formation of alkene 36 (93%, Scheme 8). Catalytic hydrogenation of 36 gave triester 37 (93%). It should be noted that during the synthesis of 37, the functional group differentiation strategy allowed us not to bother with the stereoselectivity of the transformation: the isopropylcarboxylate groups are identical, but only one of them reacted with the nucleophile leading to the corresponding bicyclic molecule possessing plane symmetry.
 | ||
| Scheme 8 Synthesis of 14·HCl. | ||
In order to install the internal nucleophile, several simple functional group transformations were performed. Since the methyl- and isopropylcarboxylate groups in 37 differ significantly in their reactivity, selective hydrolysis of the former was possible, with the formation of the carboxylic acid 38 in 88% yield. Compound 38 was transformed into acyl azide 39 via the corresponding acid chloride. The product was subjected to Curtius rearrangement without isolation; subsequent trapping of the intermediate isocyanate with tert-butanol yielded the Boc derivative 40 (88% yield from 38). Finally, deprotection of 40 with HCl in EtOAc gave 14 as its hydrochloride. Compound 14·HCl was used in the next step without purification.
In the case of 14, the key step of the synthesis – intramolecular functional group differentiation – occurred upon action of sodium methylate in refluxing methanol (Scheme 9). Due to the simultaneous transesterification, bicyclic methyl ester 41 was isolated (78% yield from 40). On the contrary, cyclization of 15, as well as (R)- or (S)-16 did not occur under these conditions and required careful optimization. We found that cyclization of 15 occurred upon treatment with trimethylaluminum and led to the formation of the bicyclic lactam 42 in 42% yield (calculating on 34, Scheme 9). Unfortunately, these conditions were unproductive in the case of 16. After some experimentation, we found that the bicyclic lactam 43 could be formed from both enantiomers of 16·HCl in 80–88% yields upon refluxing with three-fold excess of Cs2CO3 in methanol (Scheme 9). Low reactivity of 16 can be attributed to higher strain of the bicyclic system in 42.
 | ||
| Scheme 9 Intramolecular functional group differentiation in the amino diesters 14–16. | ||
Transformation sequence of the lactam 41 into the target amino acid 10 initially included reduction of 41 with borane–dimethylsulfide complex (Scheme 10). This reaction was not chemoselective and led to formation of the corresponding amino alcohol, which was isolated in 67% yield as a Boc derivative 44. For oxidation of 44, a two-step procedure was used, namely, reaction with Dess–Martin periodinane followed by Pinnick oxidation of an intermediate aldehyde 45, that gave the Boc-protected amino acid 46 (63% yield starting from 44). After deprotection of 46, the target amino acid 10 was isolated in 85% yield as the hydrochloride.
 | ||
| Scheme 10 Synthesis of the amino acid 10 (hydrochloride). | ||
In our attempts to improve the yield of the amino acid synthesis, we used an alternative approach during the final steps of synthesis of the amino acid 11 (Scheme 11). In this case, compound 42 was Boc-protected first to give 47 (quantitative yield). The amide moiety in 47 was reduced selectively in two steps and 86% overall yield via the aminal 48. Deprotection of the product 49 was performed in two steps and gave the corresponding amino acid 11 as hydrochloride.
 | ||
| Scheme 11 Synthesis of the amino acid 11 (hydrochloride). | ||
In the case of the amino acid 12, the corresponding Boc derivative (R,R)-51 was obtained in 95% yield (Scheme 12). Unfortunately, reduction of (R,R)-51 with DIBAL or LiBHEt3 was not chemoselective. Reduction of (R,R)-51 with borane–dimethylsulfide complex gave the expected alcohol (R,R)-52 in only 25–30% yield. Therefore, we turned back to the sequence described above for 10 and performed reduction of both enantiomers of 43 with borane–dimethylsulfide complex, that gave the corresponding amino alcohols (isolated as Boc derivatives 52 in 54–62% yields). Two-step oxidation of (R,R)- and (S,S)-52 under conditions used for the synthesis of 46 gave Boc-protected amino acids (R,R)- and (S,S)-53 (79–81% yield). Finally, deprotection of both enantiomers of 53 gave the target amino acids (R,R)-12 and (S,S)-12 as hydrochlorides (85–92%).
 | ||
| Scheme 12 Synthesis of enantiomers of the amino acid 12 (hydrochlorides). | ||
Optical purity of the products was checked on the alcohols 52, which were analyzed by chiral stationary phase HPLC (ChiralPack® IA column, hexane–2-propanol (97:3) as an eluent). The analyzed samples of (R,R)- and (S,S)-52 were of 97% and more than 99% ee, respectively.
Experimental part
General methods
Solvents were purified according to the standard procedures.18 Ketone 17, 9 diol 21,12 and diiodide 29 (ref. 16) were prepared using the procedures reported in the literature. All other starting materials were purchased from commercial sources. Melting points were measured on an automated melting point system. Column chromatography was performed using silica gel (230–400 mesh) as the stationary phase. 1H and 13C NMR spectra were recorded on a 500 MHz NMR spectrometer (at 499.9 MHz for protons and 124.9 MHz for carbon-13) and 400 MHz NMR spectrometer (at 400.4 MHz for protons and 100.7 MHz for carbon-13). Chemical shifts are reported in ppm downfield from TMS as an internal standard. HPLC-MS analyses were done on a LCMS instrument (chemical ionization (CI) and electrospray ionization (ESI)). GS-MS analyses were performed on a GSMS instrument with electron impact ionization (CI). Flash chromatography was performed on an automated flash chromatography system.Diisopropyl 3-(2-methoxy-2-oxoethylidene)cyclobutane-1,1-dicarboxylate (36)
To a suspension of NaH (60% in mineral oil, 4.36 g, 0.109 mol) in THF (180 mL) a solution of methyl P,P-diethylphosphonoacetate (18.2 g, 0.1 mol) in THF (90 mL) was added dropwise at 15 °C upon stirring. The mixture was stirred for 1 h, and then a solution of 17 (22.0 g, 0.091 mmol) was added. The resulting mixture was stirred at rt overnight, then most of the solvent was removed in vacuo, the residue was quenched with 10% aq NH4Cl (50 mL) and extracted with Et2O (3 × 300 mL). The combined organic phases were dried over MgSO4 and evaporated in vacuo to give 36 (25.3 g, 93%) pure enough for the next step. An analytical sample was obtained by column chromatography. Colorless oil. Rf = 0.49 (hexanes–EtOAc (5:1)). MS (m/z, CI): 299 (MH+). Anal. calc. for C15H22O6 C 60.39, H 7.43. Found C 60.18, H 7.64. 1H NMR (CDCl3) δ 5.69 (t, J = 2.0 Hz, 1H), 5.05 (sept, J = 6.2 Hz, 2H), 3.67 (s, 3H), 3.58 (d, J = 2.0 Hz, 2H), 3.30 (s, 2H), 1.22 (d, J = 6.2 Hz, 12H). 13C NMR (CDCl3) δ 170.3 (C), 166.3 (C), 157.7 (C), 114.7 (CH), 69.4 (CH), 51.2 (CH3), 49.9 (C), 41.2 (CH2), 39.1 (CH2), 21.6 (CH3).
Diisopropyl 3-(2-methoxy-2-oxoethyl)cyclobutane-1,1-dicarboxylate (37)
Ester 36 (1.0 g, 70 mmol) was dissolved in THF (140 mL), 5% Pd–C (5 g) was added, and the mixture was hydrogenated at 50 bar and 50 °C overnight. Then the catalyst was filtered off, and the filtrate was evaporated in vacuo to give 37 (19.6 g, 93%) pure enough for the next step. An analytical sample was obtained by column chromatography. Colourless oil. Rf = 0.46 (hexanes–EtOAc (5:1)). MS (m/z, CI): 301 (MH+). Anal. calc. for C15H24O6 C 59.98, H 8.05. Found C 59.67, H 7.89. 1H NMR (CDCl3) δ 5.03 (sept, J = 6.2 Hz, 1H), 4.99 (sept, J = 6.2 Hz, 1H), 3.61 (s, 3H), 2.63–2.73 (m, 3H), 2.42 (d, J = 6.7 Hz, 2H), 2.18–2.24 (m, 2H), 1.20 (d, J = 6.2 Hz, 6H), 1.18 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ 172.4 (C), 171.24 (C), 171.18 (C), 68.9 (CH), 68.8 (CH), 51.4 (CH3), 49.8 (C), 40.4 (CH2), 34.3 (CH2), 25.9 (CH), 21.6 (CH3).
[3,3-Bis(isopropoxycarbonyl)cyclobutyl]acetic acid (38)
To a solution of ester 37 (6.01 g, 20 mmol) in MeOH (100 mL), a solution of KOH (1.23 g, 22 mmol) in H2O (100 mL) and MeOH (100 mL) was added. The resulting mixture was stirred overnight, and then MeOH was removed in vacuo. The aqueous phase was washed with CH2Cl2 (3 × 100 mL), then acidified with 12 N HCl to pH = 2, and extracted with CH2Cl2 (3 × 100 mL). The organic extracts were dried over MgSO4 and evaporated to dryness to give 38 (5.07 g, 88%) pure enough for the next step. An analytical sample was obtained by column chromatography. Colourless solid. Rf = 0.65 (hexanes–EtOAc (1:2)). Mp 80–82 °C. MS (m/z, CI): 287 (MH+). Anal. calc. for C14H22O6 C 58.73, H 7.74. Found C 58.95, H 7.98. 1H NMR (CDCl3) δ 9.38 (very br s, 1H), 5.06 (sept, J = 6.1 Hz, 1H), 5.02 (sept, J = 6.1 Hz, 1H), 2.67–2.77 (m, 3H), 2.48 (d, J = 6.5 Hz, 2H), 2.22–2.27 (m, 2H), 1.22 (d, J = 6.1 Hz, 6H), 1.21 (d, J = 6.1 Hz, 6H). 13C NMR (CDCl3) δ 178.0, 171.28, 171.25, 69.00, 68.96, 49.9, 40.3, 34.3, 25.6, 21.6.
Diisopropyl 3-{[(tert-butoxycarbonyl)amino]methyl}cyclobutane-1,1-dicarboxylate (40)
To a solution of carboxylic acid 38 (4.30 g, 15 mmol) in CH2Cl2 (150 mL), oxalyl chloride (5.71 g, 45 mmol) was added at rt, the mixture was stirred at rt overnight, and then evaporated to dryness. CCl4 (2 × 100 mL) was added and evaporated; this operation was repeated twice in order to remove the residual HCl. The residue was dissolved in acetone (18 mL), and the solution was added dropwise to a solution of NaN3 (3.9 g, 60 mmol) in H2O (16 mL) at 0 °C. The mixture was stirred at 0 °C for 1.5 h, then poured into H2O (80 mL) and extracted with Et2O (4 × 50 mL). The combined organic extracts were dried over MgSO4 and evaporated to 30 mL. This solution of 39 was added dropwise to toluene (150 mL) at 105–110 °C. The mixture was stirred at this temperature for 2 h, and then tert-butanol (150 mL) was added. The resulting mixture was refluxed for 24 h and evaporated to dryness. The residue was purified by column chromatography to give 40 (4.71 g, 88%). Colourless oil. Rf = 0.26 (hexanes–EtOAc (5:1)). MS (m/z, ESI): 358 (MH+). Anal. calc. for C18H31NO6 C 60.48, H 8.74, N 3.92. Found C 60.70, H 8.46, N 4.12. 1H NMR (CDCl3) δ 5.04 (sept, J = 6.1 Hz, 1H), 5.01 (sept, J = 6.1 Hz, 1H), 4.61 (br s, 1H), 3.18 (d, J = 5.9 Hz, 0.5H), 3.12 (br s, 1.5H), 2.45–2.58 (m, 3H), 2.18–2.23 (m, 2H), 1.41 (s, 9H), 1.21 (d, J = 6.1 Hz, 6H), 1.20 (d, J = 6.1 Hz, 6H). 13C NMR (CDCl3) δ 171.4, 171.1, 156.1, 79.3, 68.93, 68.86, 49.5, 45.2, 32.0, 29.4, 28.5, 21.6.
Methyl 2-oxo-3-azabicyclo[3.1.1]heptane-1-carboxylate (41)
To a solution of 40 (7.15 g, 20 mmol) in EtOAc (40 mL), 10% HCl in EtOAc (100 mL) was added, and the mixture was stirred at rt overnight, and then evaporated to dryness. MeOH (200 mL) was added, the mixture was evaporated to dryness again. The crude 14·HCl thus obtained was dissolved in MeOH (200 mL), NaOMe (5.4 g, 0.1 mol) was added. The mixture was refluxed for 60 h and evaporated to dryness. The residue was dissolved in MeOH (200 mL), strong cation exchange resin (cationite КУ-2 resin, 100 g) was added. The mixture was stirred for 30 min, then the resin was filtered off and washed with MeOH (4 × 100 mL). The combined filtrates were evaporated to dryness, and the crude product was recrystallized from CCl4 to give 41 (2.64 g, 78%). White crystals. Mp 115–116 °C. MS (m/z, CI): 170 (MH+). Anal. calc. for C8H11NO3 C 56.80, H 6.55, N 8.28. Found C 56.53, H 6.58, N 8.52. 1H NMR (DMSO-d6) δ 7.61 (br s, 1H), 3.60 (s, 3H), 3.29 (s, 2H), 2.53–2.57 (m, 1H), 2.35–2.42 (m, 2H), 1.79–1.85 (m, 2H). 13C NMR (DMSO-d6) δ 172.6 (C), 170.9 (C), 51.5 (C), 51.4 (CH3), 44.8 (CH2), 32.7 (CH2), 27.6 (CH).tert-Butyl 1-(hydroxymethyl)-3-azabicyclo[3.1.1]heptane-3-carboxylate (44)
To a solution of 41 (2.37 g, 14 mmol) in THF (140 mL), BH3·Me2S (10 M in THF, 7 mL, 70 mmol) was added dropwise upon stirring. The resulting mixture was refluxed for 36 h, and then quenched by dropwise addition of MeOH (70 mL). The mixture was evaporated in vacuo, 10% aq NaOH (110 mL) and CH2Cl2 (50 mL) were added to the residue. The resulting mixture was stirred for 30 min, then Boc2O (4.58 g, 21 mmol) in CH2Cl2 (55 mL) was added, and the mixture was stirred vigorously at rt for 1 d. The organic phase was separated, and the aqueous phase was extracted with CH2Cl2 (3 × 100 mL). The combined organic phases were dried over MgSO4 and evaporated in vacuo. The residue was recrystallized from hexanes to give 44 (2.13 g, 67%). White crystals. Mp 97–98 °C. MS (m/z, CI): 228 (MH+). Anal. calc. for C12H21NO3 C 63.41, H 9.31, N 6.16. Found C 63.08, H 9.14, N 6.48. 1H NMR (CDCl3) δ 3.50 (br s, 2H), 3.47 (s, 2H), 3.42 (s, 2H), 2.45 (br s, 0.5H), 2.38 (br s, 0.5H), 1.90 (br s, 2H), 1.80 (br s, 1H), 1.47 (s, 9H), 1.34 (br s, 2H). 13C NMR (CDCl3) δ 156.5 (C), 79.4 (C), 67.8 and 67.4 (CH2), 52.2 and 51.8 (CH2), 50.2 and 49.8 (CH2), 42.7 (C), 32.6 and 32.4 (CH2), 28.71 (CH3), 28.67 (CH).3-(tert-Butoxycarbonyl)-3-azabicyclo[3.1.1]heptane-1-carboxylic acid (46)
Alcohol 44 (2.27 g, 10 mmol) and Dess–Martin periodinane (5.09 g, 12 mmol) were refluxed in CH2Cl2 (150 mL) for 3 h, and then stirred at rt for 12 h. The precipitate was filtered off, and the filtrate was evaporated in vacuo. The residue was subjected to flash chromatography (hexanes–EtOAc (1:1) as eluent, Rf = 0.52) to give crude 45 (2.01 g), which was dissolved in THF (65 mL); 2-methyl-2-butene (6.26 g, 89 mmol) in tert-butanol (65 mL) was added, followed by H2O (45 mL), NaClO2 (1.59 g, 18 mmol), and Na2HPO4 (4.29 g, 36 mmol). The resulting mixture was stirred at rt for 1 d, then evaporated in vacuo, diluted with H2O (50 mL), and extracted with CH2Cl2 (3 × 50 mL). The combined extracts were dried over Na2SO4 and evaporated in vacuo. The crude product was dissolved in 1 M aq NaOH (50 mL), washed with CH2Cl2 (3 × 50 mL). The aqueous phase was acidified with 1 M aq NaHSO4 to pH = 3–4 and extracted with EtOAc (3 × 100 mL). The combined extracts were dried over Na2SO4 and evaporated in vacuo to give 46 (1.53 g, 63% from 44). An analytical sample was obtained by recrystallization from benzene. White crystals. Mp 172–174 °C. MS (m/z, ESI): 242 (MH+). Anal. calc. for C12H19NO4 C 59.73, H 7.94, N 5.80. Found C 59.48, H 8.27, N 5.53. 1H NMR (CDCl3) δ 9.38 (br s, 1H), 3.65 (s, 0.8H), 3.62 (s, 1.2H), 3.47 (s, 1.2H), 3.44 (s, 0.8H), 2.32–2.41 (m, 3H), 1.53–1.55 (m, 2H), 1.42 (s, 9H). 13C NMR (CDCl3) δ 178.5 and 178.1 (C), 156.32 and 156.30 (C), 80.1 and 80.0 (C), 49.9 and 49.6 (CH2), 49.3 and 48.8 (CH2), 44.1 (C), 34.33 and 34.28 (CH2), 28.7 (CH3), 28.6 (CH).
3-Azabicyclo[3.1.1]heptane-1-carboxylic acid, hydrochloride (10·HCl)
To a solution of 46 (1.21 g, 5 mmol) in EtOAc (100 mL), 10% HCl in dioxane (50 mL) was added at rt. The mixture was stirred at rt for 4 h, then evaporated to dryness and triturated with acetone (50 mL). The precipitate was filtered and dried in vacuo to give 10·HCl (0.76 g, 85%). An analytical sample was obtained by recrystallization from EtOH–acetone. White crystals. Mp > 250 °C (dec.). Anal. calc. for C7H12ClNO2 C 47.33, H 6.81, Cl 19.96, N 7.89. Found C 47.51, H 7.07, Cl 19.90, N 7.80. 1H NMR (D2O) δ 3.69 (s, 2H), 3.53 (s, 2H), 2.60–2.68 (m, 2H), 2.57–2.61 (m, 1H), 1.82–1.87 (m, 2H). 13C NMR (D2O) δ 175.7 (C), 47.2 (CH2), 46.7 (CH2), 43.9 (C), 33.3 (CH2), 28.1 (CH).3-(tert-Butyldimethylsilyloxy)pentane-1,5-diyl dimethanesulfonate (23)
Diol 21 (139.5 g, 0.595 mol) and Et3N (195 mL, 1.40 mol) were dissolved in CH2Cl2 (0.5 L). The resulting solution was added dropwise to a solution of mesyl chloride (98.7 mL, 1.27 mol) in CH2Cl2 (1 L) at −10 to 0 °C. The resulting mixture was stirred at −10 to 0 °C for 2 h, and H2O (0.7 L) was added. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (2 × 400 mL). The combined organic phases were dried over Na2SO4, filtered, and evaporated in vacuo to give the product 23 (232 g, 100%). Yellowish crystals. Mp 61–63 °C. MS (m/z, CI): 294 (M+ − MsOH). Anal. calc. for C13H30O7S2Si C 39.98, H 7.74, S 16.42. Found C 40.36, H 7.37, S 16.05. 1H NMR (400 MHz, CDCl3) δ 4.39–4.19 (m, 4H), 4.08–3.96 (m, 1H), 2.99 (s, 6H), 2.04–1.84 (m, 4H), 0.87 (s, 9H), 0.07 (s, J = 21.3 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 65.9 (CH2), 64.8 (CH), 37.1 (CH2), 36.0 (CH3), 25.3 (CH3), 17.5 (C), −5.0 (CH3).Dimethyl 4-(tert-butyldimethylsilyloxy)cyclohexane-1,1-dicarboxylate (24)
Compound 23 (232 g, 595 mmol) and dimethyl malonate (71.3 mL, 623 mmol) were dissolved in absolute toluene (1.8 L) under argon under vigorous stirring. Lithium tert-butoxide (96.0 g, 1.20 mol) was added to the reaction mixture in portions at 0 °C. The mixture was heated at reflux overnight (effective condenser is necessary at the beginning of heating, since the reaction becomes exothermic at about 100 °C). The mixture was cooled to rt and then quenched with saturated aq NH4Cl (200 mL). After 15 min, H2O (200 mL) was added, and the layers were separated. The aqueous layer was extracted with methyl tert-butyl ether (500 mL). The combined organic extracts were dried over Na2SO4 and evaporated to give crude diester 24 (191 g), which was used without further purification. An analytical sample was obtained using flash chromatography (gradient hexanes–t-BuOMe as an eluent). Colourless liquid. MS (m/z, EI): 315 (M+ − CH3), 299 (M+ − OCH3), 273 (M+ − C4H9). Anal. calc. for C16H30O5Si C 58.15, H 9.15. Found C 58.37, H 9.30. 1H NMR (500 MHz, CDCl3) δ 3.73 (s, 3H), 3.70 (s, 3H), 2.33–2.23 (m, 2H), 1.91–1.81 (m, 2H), 1.75–1.66 (m, 2H), 1.54–1.41 (m, 2H), 0.87 (s, 9H), 0.03 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 172.0 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 171.4 (CO), 67.7 (C), 53.9 (CH), 52.1 (CH3), 52.0 (CH3), 31.1 (CH2), 27.6 (CH2), 25.4 (CH3), 17.7 (C), −5.15 (CH3), −5.19 (CH3).
O), 171.4 (CO), 67.7 (C), 53.9 (CH), 52.1 (CH3), 52.0 (CH3), 31.1 (CH2), 27.6 (CH2), 25.4 (CH3), 17.7 (C), −5.15 (CH3), −5.19 (CH3).
Dimethyl 4-hydroxycyclohexane-1,1-dicarboxylate (18)
Crude 24 (191 g, 577 mmol) was dissolved in CH3CN. KHF2 (93 g, 1.19 mol), tetrabutylammonium fluoride (1 M in THF, 58 mL, 58 mmol) and 10 mL of H2O were added to the solution. Obtained mixture was refluxed for 29 h (monitored by NMR). The mixture was cooled to rt and filtered through silica gel. The silica gel was washed thoroughly with CH3CN, and the combined filtrates were evaporated. The residue was purified by chromatography (EtOAc as an eluent) to give the product 18 (78.4 g, 61% from 23). Colourless liquid. Rf 0.54 (EtOAc). MS (m/z, EI): 198 (M+ − H2O). Anal. calc. for C10H16O5 C 55.55, H 7.46. Found C 55.27, H 7.61. 1H NMR (500 MHz, CDCl3) δ 3.71 (s, 3H), 3.67 (s, 3H), 2.35–2.25 (m, 2H), 2.13 (br s, 1H), 1.86–1.74 (m, 3H), 1.46–1.36 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 171.9 (CO), 171.2 (CO), 67.6 (CH), 53.8 (C), 52.24 (CH3), 52.16 (CH3), 30.8 (CH2), 28.1 (CH2).
Dimethyl 4-(methylsulfonyloxy)cyclohexane-1,1-dicarboxylate (32)
Alcohol 18 (78.0 g, 361 mmol) and Et3N (60.3 mL, 433 mmol) were dissolved in CH2Cl2 (400 mL). The resulting solution was added dropwise to the solution of mesyl chloride (30.7 mL, 397 mmol) in CH2Cl2 (600 mL) at −10 to 0 °C. The resulting mixture was stirred at −10 to 0 °C for 2 h, and H2O (300 mL) was added. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (2 × 200 mL). The combined organic extracts were dried over Na2SO4, filtered, and evaporated in vacuo to give 32 (104 g, 98%), which was used without further purification. An analytical sample was obtained using flash chromatography (gradient hexanes–t-BuOMe as an eluent). Colourless oil. MS (m/z, CI): 312 (MH+ + H2O), 199 (M+ − OMs). Anal. calc. for C11H18O7S C 44.89, H 6.16, S 10.89. Found C 44.62, H 5.87, S 10.74. 1H NMR (500 MHz, CDCl3) δ 4.77–4.65 (m, 1H), 3.69 (s, 3H), 3.67 (s, 3H), 2.96 (s, 3H), 2.27–2.15 (m, 2H), 2.01–1.84 (m, 4H), 1.84–1.73 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 171.0 (CO), 170.8 (CO), 77.7 (CH), 53.2 (C), 52.3 (CH3), 38.3 (CH3), 28.1 (CH2), 26.9 (CH2).
Dimethyl 4-azidocyclohexane-1,1-dicarboxylate (34)
Mesylate 32 (104 g, 481 mmol) was dissolved in a CH3CN–H2O mixture (4:1, 1 L). NaN3 (62.5 g, 961 mmol), NaI (360 mg, 2.4 mmol) and tetrabutylammonium bromide (1.55 g, 4.8 mmol) were added, and the resulting mixture was refluxed for 2 days, cooled to rt and evaporated. The residue was quenched with H2O (600 mL) and extracted with methyl tert-butyl ether (2 × 400 mL). The extracts were washed with saturated aq NaHCO3 (200 mL), dried over Na2SO4 and evaporated to give the crude azide 34 (76 g, 89%), which was used without further purification. An analytical sample was obtained using flash chromatography (gradient hexanes–t-BuOMe as an eluent). Colourless liquid. MS (m/z, CI): 214 (MH+ − N2). Anal. calc. for C10H15N3O4 C 49.79, H 6.27, N 17.42. Found C 50.07, H 6.64, N 17.38. 1H NMR (500 MHz, CDCl3) δ 3.73 (s, 3H), 3.70 (s, 3H), 3.52–3.42 (m, 1H), 2.37–2.23 (m, 2H), 1.94–1.80 (m, 4H), 1.61–1.48 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 171.4 (CO), 170.8 (CO), 57.5 (CH), 53.5 (C), 52.30 (CH3), 52.25 (CH3), 28.0 (CH2), 27.2 (CH2).
Methyl 3-oxo-2-azabicyclo[2.2.2]octane-4-carboxylate (42)
Crude azide 34 (38.0 g, 158 mmol) was dissolved in methanol (1 L), and 10% Pd on charcoal (10 g) was added. Hydrogen was bubbled through this mixture at rt until starting material disappeared (monitored by NMR; ca. 4 h). The catalyst was filtered off, and the filtrate was evaporated, dissolved in methyl tert-butyl ether (400 mL), dried over MgSO4 and evaporated again to give the amino diester 15 (32.9 g), which was used in the next step without further purification.Compound 15 (32.9 g, 154.3 mmol) was dissolved in absolute THF (1 L) under an argon atmosphere. The solution was heated to 45 °C, and a solution of AlMe3 (2 M in hexane, 95 mL, 190 mmol) was added slowly dropwise with vigorous stirring at 45–60 °C. The resulting solution was refluxed for 6 h, cooled to 0 °C, and MeOH (50 mL) was added carefully dropwise to the mixture, followed by H2O (5 mL). The resulting suspension was stirred for 1 h and filtered. The white slurry was washed with THF (4 × 250 mL) and the combined filtrates were evaporated to give crude product, which was recrystallized from benzene to give the lactam 42 (12.24 g, 42% calculating on 34). White crystals. Mp 151–152 °C. MS (m/z, CI): 184 (MH+). Anal. calc. for C9H13NO3 C 59.00, H 7.15, N 7.65. Found C 59.37, H 6.85, N 7.92. 1H NMR (500 MHz, CDCl3) δ 8.13 (br s, 1H), 3.75 (s, 3H), 3.65 (s, 1H), 2.23 (dd, J = 12.5, 9.8 Hz, 2H), 1.89–1.73 (m, 4H), 1.63 (dd, J = 17.7, 8.1 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 174.4 (CO), 171.5 (CO), 51.9 (CH), 49.9 (C), 47.0 (CH3), 26.7 (CH2), 26.3 (CH2).
2-tert-Butyl 4-methyl 3-oxo-2-azabicyclo[2.2.2]octane-2,4-dicarboxylate (47)
4-(Dimethylamino)pyridine (3.41 g, 27.9 mmol) was added to the solution of lactam 42 (10.2 g, 55.8 mmol) in acetonitrile (170 mL). A solution of Boc2O (24.3 g, 111.3 mmol) in acetonitrile (40 mL) was added over 1 h. The mixture was stirred at rt overnight and evaporated. The residue was dissolved in CHCl3 (200 mL), washed with 10% aq citric acid to pH ≈ 4 and H2O (100 mL), dried over MgSO4 and evaporated to give 47 (15.8 g, 100%). White crystals. Mp 136–138 °C. MS (m/z, CI): 228 (MH+C4H8), 184 (MH+ − C4H8–CO2). Anal. calc. for C14H21NO5 C 59.35, H 7.47, N 4.94. Found C 59.44, H 7.28, N 5.31. 1H NMR (500 MHz, CDCl3) δ 4.72 (s, 1H), 3.78 (s, 3H), 2.46–2.33 (m, 2H), 1.99–1.85 (m, 4H), 1.80–1.70 (m, 2H), 1.53 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.5 (CO), 170.3 (CO), 150.1 (CO), 83.1 (C), 52.3 (C), 52.0 (CH3), 49.8 (CH), 27.6 (CH3), 25.7 (CH2), 25.2 (CH2).
2-tert-Butyl 4-methyl 3-hydroxy-2-azabicyclo[2.2.2]octane-2,4-dicarboxylate (48)
Solution of DIBAL-H (0.7 M in toluene, 92 mL, 64.4 mmol) was added dropwise to the solution of the imide 47 (15.8 g, 55.7 mmol) in THF (600 mL) at −80 °C under an argon atmosphere. This mixture was stirred for 1 h at −80 °C, and saturated aq NH4Cl (20 mL) was added. The resulting suspension was allowed to warm to rt and stirred for additional 30 min, then filtered. The solid residue was washed thoroughly with THF (500 mL), and the combined washings were evaporated to give the N-protected hemiaminal 48 (15.0 g, 94%). White crystals. Mp 130–133 °C. MS (m/z, CI): 308 (MNa+), 212 (M+ − C4H8–OH). Anal. calc. for C14H23NO5 C 58.93, H 8.12, N 4.91. Found C 59.24, H 8.16, N 5.23. 1H NMR (400 MHz, CDCl3) δ 5.53 (s, 0.5H), 5.42 (s, 0.5H), 4.13 (s, 0.5H), 3.98 (s, 0.5H), 3.75 (s, 0.5H), 3.70 (s, 1.5H), 3.69 (s, 1.5H), 3.13 (s, 0.5H), 2.27–2.16 (m, 1H), 1.97–1.83 (m, 1H), 1.81–1.57 (m, 6H), 1.48 (s, 4.5H), 1.44 (s, 4.5H). 13C NMR (126 MHz, CDCl3) δ 174.5 and 174.3 (CO), 154.5 and 153.3 (CO), 80.4 and 79.9 (C), 79.7 and 79.5 (CH), 51.7 and 51.6 (CH3), 44.7 and 44.5 (CH), 44.2 and 43.5 (C), 28.1 and 28.0 (CH3), 27.3 and 27.0 (CH2), 25.5 and 25.3 (CH2), 24.4 and 24.0 (CH2), 19.0 (CH2).
2-tert-Butyl 4-methyl 2-azabicyclo[2.2.2]octane-2,4-dicarboxylate (49)
Solution of hemiaminal 48 (14.8 g, 51.9 mmol) and triethylsilane (8.3 mL, 51.9 mmol) in CH2Cl2 (300 mL) was cooled to −80 °C under an argon atmosphere, and BF3·Et2O (6.4 mL, 51.9 mmol) was added. This mixture was stirred for 30 min at the same temperature. The second portion of triethylsilane (8.3 mL, 51.9 mmol) was added, followed by the addition of second portion of boron trifluoride diethyl etherate (6.4 mL, 51.9 mmol). This solution was stirred for 1.5 hours and quenched with saturated aq NaHCO3 (350 mL). This mixture was stirred vigorously at rt for 20 min, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (200 mL). The combined extracts were dried over Na2SO4, evaporated and dried under vacuo to give 49 (12.8 g, 92%). White crystals. Mp 56–58 °C. MS (m/z, EI): 269 (M+), 213 (M+ − C4H8), 169 (M+ − C4H8–CO2), 57 (C4H9+). Anal. calc. for C14H23NO4 C 62.43, H 8.61, N 5.20. Found C 62.07, H 8.74, N 4.96. 1H NMR (400 MHz, CDCl3) δ 4.10 (s, 0.6H), 3.95 (s, 0.4H), 3.68 (s, 3H), 3.48 (s, 0.8H), 3.45 (s, 1.2H), 1.96–1.74 (m, 2H), 1.72–1.58 (m, 1H), 1.46 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 175.2 and 175.0 (CO), 154.3 and 154.2 (CO), 78.92 and 78.87 (C), 51.6 and 51.5 (CH3), 49.5 and 48.9 (CH2), 43.7 and 42.2 (CH), 40.1 and 40.0 (C), 28.1 (CH3), 26.9 and 26.7 (CH2), 25.6 and 25.5 (CH2).
2-(tert-Butoxycarbonyl)-2-azabicyclo[2.2.2]octane-4-carboxylic acid (50)
A solution of NaOH (2 M in H2O, 55.7 mL, 111 mmol) was added to a solution of ester 49 (12.0 g, 44.6 mmol) in MeOH (150 mL). The mixture was stirred at rt overnight and evaporated. The residue was dissolved in H2O (150 mL), washed with Et2O (2 × 100 mL), acidified to pH = 3 by addition of 1 M aq NaHSO4 and extracted with EtOAc (3 × 150 mL). The combined extracts were dried over Na2SO4 and evaporated to give 50 (10.4 g, 91%). White crystals. Mp 162–163 °C. MS (m/z, CI): 200 (MH+ − C4H8), 156 (MH+ − C4H8–CO2); 254 (M − H+). Anal. calc. for C13H21NO4 C 61.16, H 8.29, N 5.49. Found C 60.89, H 8.02, N 5.27. 1H NMR (400 MHz, CDCl3) δ 9.12 (br s, 1H), 4.10 (s, 0.7H), 3.95 (s, 0.3H), 3.55–3.39 (m, 2H), 1.97–1.75 (m, 6H), 1.75–1.56 (m, 2H), 1.44 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 180.4 and 180.2 (CO), 154.81 and 154.77 (CO), 79.7 and 79.6 (C), 49.7 and 49.0 (CH2), 44.1 and 42.7 (CH), 40.3 and 40.2 (C), 28.5 (CH3), 27.2 and 27.0 (CH2), 25.9 and 25.8 (CH2).
2-Azabicyclo[2.2.2]octane-4-carboxylic acid, hydrochloride (11·HCl)
Saturated HCl in dioxane (150 mL) was added to the solution of 50 (6.68 g, 26.2 mmol) in 250 mL of EtOAc. The mixture was stirred overnight and filtered. The solid was dried in vacuo to give the hydrochloride of 11 (4.11 g, 82%). White crystals. Mp > 250 °C (dec.). Anal. calc. for C8H14ClNO2 C 50.14, H 7.36, Cl 18.50, N 7.31. Found C 49.86, H 7.03, Cl 18.73, N 6.99. 1H NMR (400 MHz, DMSO-d6) δ 12.82 (br s, 1H), 9.22 (br s, 2H), 3.41 (s, 1H), 3.13 (s, 2H), 2.04–1.90 (m, 2H), 1.88–1.62 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 175.2 (CO), 44.0 (CH2), 43.6 (CH), 36.4 (C), 25.6 (CH2), 21.4 (CH2).
Dimethyl (3S)-3-(benzyloxy)cyclopentane-1,1-dicarboxylate (30)
To a suspension of NaH (14,4 g, 0.360 mol, 60% in mineral oil) in THF (840 mL) a solution of dimethyl malonate (50.0 g, 0.378 mol) in THF (840 mL) was added dropwise at rt with stirring under an argon atmosphere. The resulting mixture was stirred for 1 h, then (2S)–O-benzyl-1,4-diiodobutan-2-ol (29, 50.0 g, 0.120 mol) in THF (50 mL) was added slowly. The resulting mixture was refluxed for 12 h, then cooled, quenched with saturated aq NH4Cl (100 mL) and stirred for 15 min. Most of THF was removed in vacuo, H2O (400 mL) was added, and the product was extracted with CH2Cl2 (3 × 400 mL). The combined extracts were dried over Na2SO4, filtered and evaporated in vacuo to give the crude product (ca. 75 g) containing an excess of dimethyl malonate. To obtain pure 30, multiple chromatographic purifications were performed (hexanes–EtOAc (5:1) as an eluent). Colourless oil. [α]20D +12.4 (c 3.2, CHCl3). MS (m/z, EI): 186 (M+ − C6H5CHO), 145, 91 (C7H7+). Anal. calc. for C16H20O5 C 65.74, H 6.90. Found C 65.58, H 7.06. 1H NMR (500 MHz, CDCl3) δ 7.24–7.34 (m, 5H), 4.47 (d, J = 11.8 Hz, 1H), 4.42 (d, J = 11.8 Hz, 1H), 4.08 (m, 1H), 3.71 (s, 3H), 3.70 (s, 3H), 2.43–2.58 (m, 3H), 2.11 (m, 1H), 1.87–s1.92 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 173.1, 172.4, 138.6, 128.4, 127.63, 127.57, 79.7, 70.7, 59.1, 52.9, 52.8, 40.1, 32.0, 31.9.
Dimethyl (3S)-3-hydroxycyclopentane-1,1-dicarboxylate ((S)-19)
The crude 30 obtained in the previous experiment (ca. 75 g) was dissolved in MeOH (800 mL), and 5% Pd–C (40 g) was added. The mixture was hydrogenated in an autoclave at 70 °C and 100 atm of H2 for 3 d. The catalyst was filtered off, the solvent was removed in vacuo, and the residue was purified by chromatography (hexanes–EtOAc (2:1 to 1:1) as an eluent) to give the product (S)-19 (18.8 g, 76% over 2 steps). Colourless oil. [α]20D −4.7 (c 0.8, CHCl3). MS (m/z, CI): 203 (MH+). Anal. calc. for C9H14O5 C 53.46, H 6.98. Found C 53.21, H 7.37. 1H NMR (500 MHz, CDCl3) δ 4.36–4.40 (m, 1H), 3.74 (s, 3H), 3.71 (s, 3H), 2.41–2.47 (m, 1H), 2.31–2.39 (m, 2H), 2.22–2.28 (m, 1H), 2.02 (br s, 1H), 1.87–1.95 (m, 1H), 1.72–1.79 (m, 1H). 13C NMR (125 MHz, CDCl3) δ 173.8, 172.9, 73.3, 59.2, 53.2, 53.0, 43.6, 35.3, 32.3.
Dimethyl (3S)-3-[(methylsulfonyl)oxy]cyclopentane-1,1-dicarboxylate ((S)-33)
Alcohol (S)-19 (8.02 g, 39.5 mmol) and Et3N (5.95 g, 55.3 mmol) were dissolved in CH2Cl2 (120 mL). The solution was cooled to −10 °C, and mesyl chloride (5.44 g, 47.5 mmol) was added dropwise. The resulting mixture was stirred at −10 to 0 °C for 3 h, and H2O (100 mL) was added. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (2 × 100 mL). The combined organic phases were dried over Na2SO4, filtered, and evaporated in vacuo to give the product (S)-33 (11.1 g, 99%). Colourless oil. [α]20D +12.1 (c 1.0, CHCl3). MS (m/z, CI): 303 (MNa+), 185 (M+ − OMs). Anal. calc. for C10H16O7S C 42.85, H 5.75, S 11.44. Found C 42.83, H 6.10, S 11.15. 1H NMR (500 MHz, CDCl3) δ 5.19 (m, 1H), 3.75 (s, 3H), 3.72 (s, 3H), 2.99 (s, 3H), 2.59–2.69 (m, 2H), 2.49–2.55 (m, 1H), 2.20–2.25 (m, 1H), 2.01–2.13 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 172.2, 171.6, 82.6, 58.8, 53.2, 53.2, 41.1, 38.8, 33.1, 31.9.Dimethyl (3R)-3-azidocyclopentane-1,1-dicarboxylate ((R)-35)
Mesylate (S)-33 (11.1 g, 39.5 mmol) was dissolved in MeCN (110 mL), NaN3 (15.4 g, 0.237 mol) was added, and the resulting mixture was refluxed for 8 d, then cooled, filtered through silica gel, and evaporated in vacuo to give the product (R)-35 (8.81 g, 98%). Colourless oil. [α]20D −8.4 (c 1.58, CHCl3). MS (m/z, EI): 199 (M+ − N2), 171, 127, 113. Anal. calc. for C9H13N3O4 C 47.57, H 5.77, N 18.49. Found C 47.38, H 5.54, N 18.27. 1H NMR (500 MHz, CDCl3) δ 4.03–4.07 (m, 1H), 3.74 (s, 3H), 3.72 (s, 3H), 2.53–2.57 (m, 1H), 2.44–2.47 (m, 1H), 2.30–2.33 (m, 1H), 2.15–2.21 (m, 1H), 1.92–2.00 (m, 1H), 1.76–1.83 (m, 1H). 13C NMR (125 MHz, CDCl3) δ 172.5, 171.9, 62.0, 58.9, 53.1, 53.1, 39.9, 32.3, 31.8.Dimethyl (3R)-3-aminocyclopentane-1,1-dicarboxylate, hydrochloride ((R)-16·HCl)
To a solution of (R)-35 (8.81 g, 38.7 mmol) in MeOH (150 mL), 5% Pd–C (6 g) and saturated HCl in dioxane (30 mL) were added. A steam of hydrogen was passed through the resulting mixture with stirring for 6 h. The catalyst was filtered off, and the solvent was removed in vacuo to give the product (R)-16·HCl (8.74 g, 95%). White powder. Mp 138–139 °C. [α]20D +12.1 (c 1.0, MeOH). MS (m/z, CI): 202 (MH+). Anal. calc. for C9H16ClNO4 C 45.48, H 6.79, Cl 14.92, N 5.89. Found C 45.62, H 7.06, Cl 15.13, N 6.02. 1H NMR (500 MHz, DMSO-d6) δ 8.47 (br s, 3H), 3.66 (s, 3H), 3.65 (s, 3H), 3.40–3.50 (m, 1H), 2.58–2.62 (m, 1H), 2.31–2.36 (m, 1H), 1.97–2.14 (m, 3H), 3.67–1.75 (m, 1H). 13C NMR (125 MHz, DMSO-d6) δ 171.4, 171.0, 58.4, 53.0, 52.9, 50.0, 37.6, 31.3, 29.6.Methyl (1R,4R)-3-oxo-2-azabicyclo[2.2.1]heptane-4-carboxylate ((R,R)-43)
Compound (R)16·HCl (3.01 g, 17.3 mmol) was dissolved in MeCN (170 mL), and Cs2CO3 (16.9 g, 51.8 mmol) was added. The mixture was refluxed with stirring for 6 h and then cooled. Saturated HCl in dioxane (100 mL) was added, and the mixture was stirred for 30 min, then filtered through silica gel. The filtrates were evaporated in vacuo to give the product (1R,4R)-43 (2.10 g, 72%) pure enough for the next step. An analytical sample was obtained by gradient chromatography (hexanes–t-BuOMe to MeCN). Colourless oil. [α]20D +84.4 (c 1.0, CHCl3). MS (m/z, CI): 170 (MH+). Anal. calc. for C8H11NO3 C 56.80, H 6.55, N 8.28. Found C 56.48, H 6.31, N 8.02. 1H NMR (500 MHz, CDCl3) δ 6.78 (br s., 1H), 3.89 (s, 1H), 3.80 (s, 3H), 2.23–2.27 (m, 1H), 2.13 (m, 1H), 1.81–1.96 (m, 2H), 1.71–1.76 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 176.8, 170.5, 59.2, 54.4, 52.5, 44.6, 31.4, 26.6.2-tert-Butyl 4-methyl (1R,4R)-3-oxo-2-azabicyclo[2.2.1]heptane-2,4-dicarboxylate ((R,R)-51)
Bicyclic lactam (1R,4S)-43 (2.02 g, 11.8 mmol) was dissolved in MeCN (50 mL), and dimethylaminopyridine (DMAP, 0.14 g, 1.2 mmol) was added, followed by Boc2O (3.87 g, 17.7 mmol). The mixture was stirred at rt for 4 h. The solvent was removed in vacuo, and the residue was purified by column chromatography (hexanes–EtOAc (2:1)) to give the product (1R,4S)-51 (2.86 g, 90%). Yellowish powder. Mp 93–94 °C. [α]20D −5.2 (c 1.0, CHCl3). MS (m/z, CI): 292 (MNa+), 214 (MH+ − C4H8). Anal. calc. for C13H19NO5 C 57.98, H 7.11, N 5.20. Found C 58.25, H 7.36, N 4.83. 1H NMR (400 MHz, CDCl3) δ 4.53–4.57 (m, 1H), 3.80 (s, 3H), 2.17–2.31 (m, 2H), 1.96–2.06 (m, 2H), 1.85–1.92 (m, 1H), 1.76 (d, J = 10.2 Hz, 2H), 1.52 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 170.7, 169.2, 149.5, 83.5, 61.0, 57.7, 52.6, 41.2, 29.5, 28.2, 26.9.
tert-Butyl (1R,4R)-4-(hydroxymethyl)-2-azabicyclo[2.2.1]heptane-2-carboxylate ((R,R)-52)
Prepared from (1R,4R)-43 using the method described above for the synthesis of 44, except the purification was done by column chromatography (hexanes–EtOAc (1:1) as an eluent). Yield 1.62 g (62%). Colourless oil. [α]20D –51.9 (c 1.0, MeOH). MS (m/z, EI): 227 (M+), 171 (M+ − C4H8), 140, 96, 57 (C4H9+). Anal. calc. for C12H21NO3 C 63.41, H 9.31, N 6.16. Found C 63.34, H 8.95, N 6.37. 1H NMR (500 MHz, CDCl3) δ 4.22 (s, 0.5H), 4.11 (s, 0.5H), 3.73 (s, 2H), 3.21 (m, 1H), 2.98 (m, 1H), 2.66–2.87 (br, 1H), 1.54–1.69 (m, 4H), 1.41 (m, 9H), 1.29–1.42 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 154.6, 154.3, 79.3, 79.1, 64.6, 64.4, 58.0, 57.0, 55.0, 54.5, 51.1, 50.7, 40.1, 39.8, 31.4, 31.2, 30.2, 29.9, 28.7.
(1R,4R)-2-(tert-Butoxycarbonyl)-2-azabicyclo[2.2.1]heptane-4-carboxylic acid ((R,R)-53)
Prepared from (1R,4R)-52 using the method described above for the synthesis of 46. Yield 1.35 g (79%). White powder. Mp 129–130 °C. [α]20D −41.5 (c 1.0, CHCl3). MS (m/z, CI): 242 (MH+). Anal. calc. for C12H19NO4 C 59.73, H 7.94, N 5.80. Found C 60.09, H 7.68, N 6.14. 1H NMR (500 MHz, CDCl3) δ 4.36 (s, 0.5H), 4.22 (s, 0.5H), 3.55 (s, 1H), 3.21–3.31 (m, 1H), 2.11 (m, 1H), 1.93 (m, 1H), 1.75–1.81 (m, 4H), 1.45 (m, 9H); COOH is exchanged with HDO. 13C NMR (125 MHz, CDCl3) δ 178.3, 154.3 and 154.0, 79.8, 58.3 and 57.4, 54.2 and 53.8, 52.7 and 52.2, 41.5 and 41.2, 31.7 and 31.4, 31.2 and 30.9, 28.5.(1R,4R)-2-Azabicyclo[2.2.1]heptane-4-carboxylic acid, hydrochloride ((R,R)-12·HCl)
To a solution of the compound (1R,4R)-53 (70 mg, 0.30 mmol) in EtOAc (10 mL), saturated HCl in dioxane (5 mL) was added. The solution was stirred at rt for 12 h, then evaporated to dryness and triturated with acetone (5 mL). The precipitate was filtered and dried in vacuo to give the product (1R,4R)-12·HCl (49 mg, 92%). White powder. Mp > 250 °C (dec.). [α]20D +91.0 (c 1.0, D2O). Anal. calc. for C7H12ClNO2 C 47.33, H 6.81, Cl 19.96, N 7.89. Found C 47.64, H 7.10, Cl 19.74, N 8.16. 1H NMR (500 MHz, D2O) δ 4.15 (s, 1H), 3.42 (d, J = 10.7 Hz, 1H), 3.25 (d, J = 10.7 Hz, 1H), 1.81–2.15 (m, 6H). 13C NMR (125 MHz, D2O) δ 175.5, 59.3, 51.6, 50.8, 39.9, 30.5, 25.1.Dimethyl (3R)-3-[(4-nitrobenzoyl)oxy]cyclopentane-1,1-dicarboxylate (31)
To a pre-cooled solution (−10 °C) of PPh3 (19.4 g, 74.2 mmol) in THF (150 mL), dimethyl azodicarboxylate (12.9 g, 74.2 mmol) was added dropwise. The mixture was stirred for 30 min, and (S)-19 (10.0 g, 49.5 mmol) and 4-nitrobenzoic acid (12.4 g, 74.2 mmol) in THF (100 mL) was added. The resulting mixture was stirred at rt overnight, and the solvent was removed in vacuo. The residue was subjected to chromatography (hexanes–EtOAc (2:1)) to give the product 31 (12.3 g, 71%). White powder. [α]20D −48.1 (c 1.0, CHCl3). MS (m/z, CI): 351 (M − H+); 370 (MH+ + H2O), 185 (M+ − 4-O2NC6H4COO). Anal. calc. for C16H17NO8 C 54.70, H 4.88, N 3.99. Found C 55.03, H 5.16, N 3.90. 1H NMR (500 MHz, CDCl3) δ 8.27 (d, 2H), 8.13 (d, 2H), 5.51 (m, 1H), 3.75 (s, 3H), 3.70 (s, 3H), 2.58–2.74 (m, 3H), 2.30 (m, 1H), 2.05–2.15 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 172.5, 172.2, 164.2, 150.8, 135.8, 130.9, 123.7, 77.7, 59.3, 53.2, 53.1, 40.8, 32.3, 32.3.
Dimethyl (3R)-3-hydroxycyclopentane-1,1-dicarboxylate ((R)-19)
To a solution of 31 (9.14 g, 26 mmol) in MeOH (150 mL), K2CO3 (1.8 g, 13 mmol) was added in one portion. The mixture was stirred at rt for 2 h, then evaporated to dryness. The residue was subjected to column chromatography (hexanes–EtOAc (1:1)) to give the product (R)-19 (4.98 g, 95%). Colourless oil. [α]20D + 4.2 (c 0.8, CHCl3). Other spectroscopic and physical characteristics were identical to those of (S)-19.
Dimethyl (3R)-3-[(methylsulfonyl)oxy]cyclopentane-1,1-dicarboxylate ((R)-33)
Prepared from (R)-19 analogously to the enantiomer. Yield 96%. Colourless oil. [α]20D −12.6 (c 1.0, CHCl3). Other spectroscopic and physical characteristics were identical to those of (S)-33.Dimethyl (3S)-3-azidocyclopentane-1,1-dicarboxylate ((S)-35)
Prepared from (R)-33 analogously to the enantiomer. Yield 93%. Colourless oil. [α]20D + 7.8 (c 1.56, CHCl3). Other spectroscopic and physical characteristics were identical to those of (R)-35.Dimethyl (3S)-3-aminocyclopentane-1,1-dicarboxylate, hydrochloride ((S)-16·HCl)
Prepared from (S)-35 analogously to its enantiomer. Yield 88%. White powder. Mp 141–142 °C. [α]20D −12.6 (c 1.0, MeOH). Other spectroscopic and physical characteristics were identical to those of (R)-16·HCl.Methyl (1S,4S)-3-oxo-2-azabicyclo[2.2.1]heptane-4-carboxylate ((S,S)-43)
Prepared from (S)16·HCl analogously to its enantiomer. Yield 64%. Colourless oil. [α]20D −81.1 (c 1.0, CHCl3). Other spectroscopic and physical characteristics were identical to those of (R,R)-43.tert-Butyl (1S,4S)-4-(hydroxymethyl)-2-azabicyclo[2.2.1]heptane-2-carboxylate ((S,S)-52)
Prepared from (S,S)-43 analogously to its enantiomer. Yield 54%. Colourless oil. [α]20D +47.1 (c 1.0, MeOH). Other spectroscopic and physical characteristics were identical to those of (1R,4R)-52.(1S,4S)-2-(tert-Butoxycarbonyl)-2-azabicyclo[2.2.1]heptane-4-carboxylic acid ((S,S)-53)
Prepared from (S,S)-53 analogously to its enantiomer. Yield 81%. White powder. Mp 131–133 °C. [α]20D +46.4 (c 1.0, CHCl3). Other spectroscopic and physical characteristics were identical to those of (R,R)-53.(1S,4S)-2-Azabicyclo[2.2.1]heptane-4-carboxylic acid, hydrochloride ((S,S)-12·HCl)
Prepared from (S,S)-53 analogously to its enantiomer. Yield 85%. White powder. Mp > 250 °C (dec.). [α]20D −85.4 (c 1.0, D2O). Other spectroscopic and physical characteristics were identical to those of (R,R)-12·HCl.Conclusions
Intramolecular functional group differentiation in cyclic amino diesters 14–16 proved to be an efficient strategy for practical synthesis of bicyclic β-amino acids, analogues of the nipecotic acid 10–12. Proper positioning of the nucleophilic amino group relatively to the two carboxylates during retrosynthetic analysis of the target molecules resulted in completely regioselective key lactam formation. Conditions of the key transformation, as well as subsequent reduction of the lactam moiety were optimized. Our results as well as literature data show that the strategy based on intramolecular functional group differentiation can be considered as general and effective approach to the construction of the bicyclic scaffolds.Acknowledgements
The authors thank Ms Kseniya Krasnopolska for the chromatographic separations, Vitaly Polovinko for NMR spectra measurements and Valentin Tkachenko for LC – MS measurements.Notes and references
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Nat. Chem., 2012, 4, 90–98 CrossRef CAS PubMed
.
- O. O. Grygorenko, D. S. Radchenko, D. M. Volochnyuk, A. A. Tolmachev and I. V. Komarov, Chem. Rev., 2011, 111, 5506–5568 CrossRef CAS PubMed
-
(a) J. Vagner, H. Qu and V. Hruby, Curr. Opin. Chem. Biol., 2008, 12, 292–296 CrossRef CAS PubMed
-
(a) P. Spiteller and F. von Nussbaum, in Enantioselective Synthesis of β-Amino Acids, ed. E. Juaristi and V. A. Soloshonok, John Wiley and Sons, Hoboken, NJ, 2nd edn, 2005, pp. 19–91 Search PubMed
- For selected examples, see:
(a) S. Kotha, D. Goyal and A. S. Chavan, J. Org. Chem., 2013, 78, 12288–12313 CrossRef CAS PubMed
-
(a) Y. M. Ivon, A. V. Tymtsunik, I. V. Komarov, O. V. Shishkin and O. O. Grygorenko, Synthesis, 2015, 47, 1123–1130 CrossRef CAS
-
(a) D. Casabona, A. I. Jiménez and C. Cativiela, Tetrahedron, 2007, 63, 5056–5061 CrossRef CAS PubMed
- The preliminary results of this work were published as a letter, see: A. V. Tymtsunik, V. A. Bilenko, O. O. Grygorenko and I. V. Komarov, Synlett, 2014, 25, 355–358 CAS
- D. S. Radchenko, P. K. Mykhailiuk, I. V. Komarov and A. V. Bezdudny, Synlett, 2009, 1827–1829 CAS
-
(a) D. R. Walley and J. L. Belletire, Synth. Commun., 1984, 14, 401–406 CrossRef CAS
- While the work was in progress, a more convenient approach to 17 was described: S. B. Singh, D. E. Kaelin, J. Wu, L. Miesel, C. M. Tan, P. T. Meinke, D. Olsen, A. Lagrutta, P. Bradley, J. Lu, S. Patel, K. W. Rickert, R. F. Smith, S. Soisson, C. Wei, H. Fukuda, R. Kishii, M. Takei and Y. Fukuda, ACS Med. Chem. Lett., 2014, 5, 609–614 CrossRef CAS PubMed
-
(a) J. A. C. Romero, S. A. Tabacco and K. A. Woerpel, J. Am. Chem. Soc., 2000, 122, 168–169 CrossRef CAS
-
(a) R. Babu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116, 986–997 CrossRef
- W. Tang, X. Wei, N. K. Yee, N. Patel, H. Lee, J. Savoie and C. H. Senanayake, Org. Process Res. Dev., 2011, 15, 1207–1211 CrossRef CAS
- D. Ma, J. Ma and L. Dai, Tetrahedron: Asymmetry, 1997, 8, 825–827 CrossRef CAS
- S. P. Chavan and P. B. Lasonkar, Tetrahedron: Asymmetry, 2012, 23, 1496–1500 CrossRef CAS
- P. Gmeiner and D. Junge, J. Org. Chem., 1995, 60, 3910–3915 CrossRef CAS
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Elsevier, Oxford, 2003, pp. 1–609 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra and CSP HPLS traces. See DOI: 10.1039/c6ra01548d |
| This journal is © The Royal Society of Chemistry 2016 |