
A multi-stimuli-responsive fluorescence switch based on E–Z isomerization of hydrazone†
Hai-Rong Zhengac,
Li-Ya Niu*ab,
Yu-Zhe Chena,
Li-Zhu Wua,
Chen-Ho Tunga and
Qing-Zheng Yangb
aTechnical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: niuly@mail.ipc.ac.cn; Tel: +86 10 82543462
bCollege of Chemistry, Beijing Normal University, Beijing 100875, China
cUniversity of the Chinese Academy of Sciences, Beijing 100049, China
First published on 18th April 2016
Abstract
We report a new strategy to construct fluorescence switches by taking advantage of the E–Z isomerization of the hydrazone group. The switch, synthesized by linking pyridyl-2-aldehydehydrazone to a BODIPY fluorophore, was able to exist in three forms: E-BODIPY, Z-BODIPY and protonated BODIPY with different photophysical properties. Reversible ON/OFF fluorescence switching among them was realized by multiple stimuli such as irradiation and addition of acid/base.
Multi-stimuli-responsive fluorescence switches have attracted increasing interest for their potential applications in developing data storage materials,1,2 sensors,3–5 and logic gates.6 Such switches are usually obtained by attaching a stimuli-responsive group to a fluorophore to alter its fluorescence.7–12 Boron dipyrromethene (BODIPY) dyes present outstanding chemistry and spectroscopic properties, such as high molar extinction coefficients, high fluorescence quantum yield, insensitivity to solvent polarity and pH, thermal and photochemical stability.13,14 Furthermore, the versatility of synthetic strategies of BODIPYs allows the creation of a perfect fit between the structure of the dyes and their desired photophysical properties, making BODIPYs attractive for conducting required applications,15–22 including fluorescence switches. Several fluorescent switches using BODIPY as fluorophore have been synthesized and studied, most of them were based on Förster resonance energy transfer (FRET) or photo-induced electron transfer (PET) to generate ON/OFF behaviour of BODIPY dyes.23–27 The Neckers group23 attached photochromic dithienylethene moieties to fluorescent BODIPY covalently via a phenylacetylene linker. The photo-responsive switches were highly emissive as open-ring isomers; while their fluorescence was significantly quenched in closed-ring isomers. Daub et al.24 developed a distinctly controllable and reversible fluorescent switch based on the flavin group, one of the most important biological redox-active dye units. The oxidized and reduced forms of the dyad were switched the by various input signals, such as photo or thermal activated thiol/disulfide redox chemistry, which tuned the efficiency of the electron transfer process.
In this work, we report a new strategy to construct multi-stimuli-responsive fluorescence switch by taking advantage of the E–Z isomerization of the hydrazone group. The hydrazone functional group is versatile and has found extensive applications in various fields,28 such as dynamic combinatorial chemistry,29,30 foldamers,31,32 molecular switches,33–35 and sensors.36,37 One important feature of the hydrazone groups, upon appropriate substitution, is that they can undergo configurational changes under UV irradiation, as a result of photoinduced rotation around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond.38 Recently, Aprahamian et al. showed that configurational changes can also be accomplished with pH and discussed the mechanism of pH induced configurational switching.39 Then they employed the hydrazone functional group in the development of light activated and chemically controllable switch, which was used to control the photophysical properties of liquid crystals.40 Besides, they have converted the hydrazine into light emitting materials,41,42 visible-light activated azo switches43 and chemical sensors.44 However, fluorescent switches based on E–Z isomerization of hydrazone have been rarely reported. Herein, we linked pyridyl-2-aldehydehydrazone to a BODIPY fluorophore and realized the “visualization” of the E–Z isomerization. The switch exists in three forms: E-BODIPY, Z-BODIPY and protonated BODIPY with different photophysical properties. Reversible ON/OFF fluorescence switching among them was achieved by multiple stimuli such as irradiation and addition of acid/base (Scheme 1).
N bond.38 Recently, Aprahamian et al. showed that configurational changes can also be accomplished with pH and discussed the mechanism of pH induced configurational switching.39 Then they employed the hydrazone functional group in the development of light activated and chemically controllable switch, which was used to control the photophysical properties of liquid crystals.40 Besides, they have converted the hydrazine into light emitting materials,41,42 visible-light activated azo switches43 and chemical sensors.44 However, fluorescent switches based on E–Z isomerization of hydrazone have been rarely reported. Herein, we linked pyridyl-2-aldehydehydrazone to a BODIPY fluorophore and realized the “visualization” of the E–Z isomerization. The switch exists in three forms: E-BODIPY, Z-BODIPY and protonated BODIPY with different photophysical properties. Reversible ON/OFF fluorescence switching among them was achieved by multiple stimuli such as irradiation and addition of acid/base (Scheme 1).
 | ||
| Scheme 1 Conversion of Z-BODIPY, E-BODIPY and protonated-BODIPY. | ||
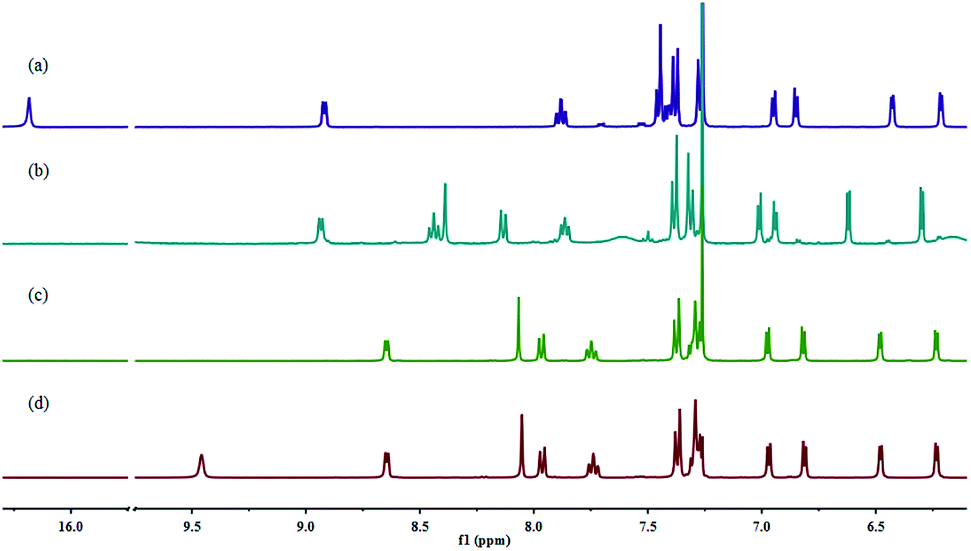
It is quite convenient to introduce substituents on the 3- and 5-positions of the 3,5-dichloro-BODIPY via nucleophilic substitution.45 Treatment of 3,5-dichloro-BODIPY with hydrazine hydrate as nucleophile in methanol at room temperature easily afforded BODIPY-hydrazine.46 After the reaction of BODIPY-hydrazine with pyridyl-2-aldehyde, both E and Z isomers were obtained and easily isolated through column chromatography. It is noteworthy that the two forms showed high stability under different conditions in CHCl3 (day light, high temperature) (Fig. S1†). As shown in Fig. 1, the 1H NMR spectra of E- and Z-BODIPY were distinct from each other. An obvious NH signal was observed at 16.17 ppm for the Z configuration, indicating that an intramolecular H-bond between the pyridine and the hydrazone NH was formed. By contrast, signal of the exchangeable NH in E-BODIPY, which was broader than that of Z form, was observed at 9.45 ppm.
 | ||
| Fig. 1 Partial 1H-NMR spectra of Z-BODIPY and E-BODIPY in CDCl3. | ||
We studied the absorption and emission spectra of both isomers. The UV-vis spectra of the two isomers were similar, except that the Z-BODIPY showed a little red shift of about 7 nm (Fig. 2). E-BODIPY emitted strong fluorescence at 570 nm, which showed obvious red shift compared to that of BODIPY-hydrazine (525 nm (ref. 46)). It was in accordance with the formation of other aromatic hydrazone and aliphatic hydrazone.46 In contrast, the Z-BODIPY was almost non-fluorescent. The fluorescence quenching was also observed in other hydrogen-bonded aromatic systems. According to literatures, the quenching of fluorescence might be attributed to the charge-transfer (CT) interaction in the intramolecular hydrogen-bonding system (Scheme 2).47–49 In the Z-BODIPY molecule, the NH of hydrazine group acted as proton-donating substituent, while the pyridine group acted as the proton-accepting substituent. At ground state, there was an intramolecular hydrogen bond between the proton-donating and proton-accepting substituents. Once excited, rapid electron transfer took place along the hydrogen bond with the formation of a highly polarized charge transfer (CT) state. Then the electron transfer back rapidly to the ground state in a non-radiative way, thereby resulting in the fluorescence quenching.49
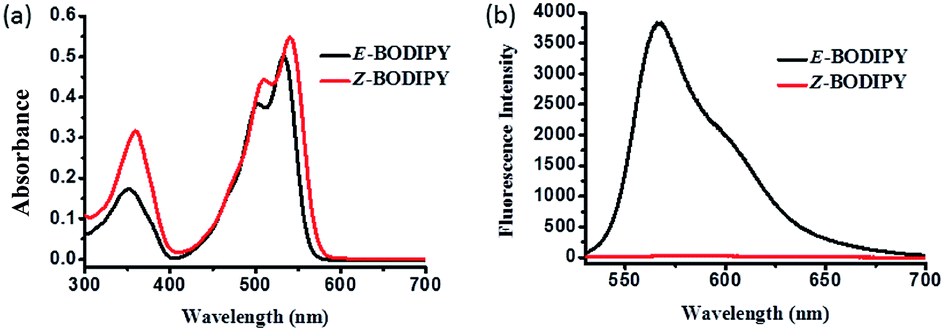 | ||
| Fig. 2 (a) Absorption and (b) fluorescence spectra of the E- and Z-BODIPY (10 μM) in dichloromethane. λex = 520 nm. | ||
 | ||
| Scheme 2 Charge-transfer (CT) interaction in Z-BODIPY between hydrazine group and the pyridine group. Abbreviations: ET, electron transfer; CT, charge transfer state; IC, internal conversion. | ||
The CN bond of pyridyl-2-aldehydehydrazone subunit in E-BODIPY underwent E–Z photoisomerization upon UV light irradiation. By contrast, the Z isomer is highly resistant to the photoirradiation as a result of the intramolecular H-bond.28 We used fluorescence spectra to monitor the photoisomerization process of E-BODIPY to Z-BODIPY. A solution of E-BODIPY (10 μM) in CH2Cl2 was irradiated in a quartz cuvette with high-pressure Hg lamp (500 W) with a filter (300 nm < λ < 400 nm). As a result the fluorescence intensity gradually decreased upon the irradiation (Fig. 3). In addition, this process was monitored by HPLC, which showed that the photoconversion ratio of E to Z isomer was about 60% after irradiated (Fig. S2†). The photoisomerization of E-BODIPY to Z-BODIPY was further identified by 1H NMR. After the irradiation of E-BODIPY (20 mM) in CD2Cl2 in a glass tube, the N–H proton signal was shifted from 9.45 to 16.17 ppm. This significant down-field shift indicated that the hydrogen bond formed between N atom of pyridine group and the NH proton, suggesting the formation of Z-BODIPY. The photoconversion of E to the Z isomer resulted in 67% yield according to the ratio of the integration, which is in accordance with the HPLC analyses (Fig. S3†). Quantum yield for E to Z isomerization under UV-irradiation of E-BODIPY was calculated to be 0.41 (see the ESI for details†), which is in good agreement with literature values.50
 | ||
| Fig. 3 Fluorescence spectra of the E-BODIPY (10 μM) dichloromethane upon irradiation with high-pressure Hg lamp (500 W) with a filter (300 nm < λ < 400 nm). λex = 520 nm. | ||
In addition to the photo-responsive properties, the ON/OFF behaviour of BODIPY dye was also achieved with treatment of acid/base. We used fluorescence and UV/vis spectroscopy to monitor this process. As shown in the fluorescence spectra (Fig. 4), the fluorescence of E-BODIPY was quenched after addition of trifluoroacetic acid. At the same time, a bathochromic shift of about 20 nm was observed in the UV/vis spectra (Fig. S4†), accompanied with a colour change from pink to purple (Fig. S6†). The product was identified with 1H NMR as the protonated BODIPY. As shown in Fig. 5, the protons of pyridine group in the protonated BODIPY shifted downfield, as a result of the protonation of the pyridine group. On the other hand, the addition of trifluoroacetic acid to the Z-BODIPY led to the same protonated product with E-BODIPY, as identified by both absorption and 1H NMR spectra. We proposed that trifluoroacetic acid broke the H-bond and protonated the nitrogen of the pyridine group in Z-BODIPY, leading to the rotation around the CN double bond and resulted in an E-form with smaller steric hindrance than the Z-form. The Z → E isomerization process involved a polar transition state of hydrazone-azo tautomerization followed by rotation around a C–N single bond (Scheme 3).36,51–53 The hydrazone-azo tautomerization also explained the bathochromic shift in the absorption band and the fluorescence quenching. Furthermore, after the addition of N,N-diisopropylethylamine (DIPEA) to the solution of protonated BODIPY, the fluorescence enhancement at 570 nm was observed, as shown in Fig. 4b. 1H NMR spectra of the protonated BODIPY after adding base was identical to the E-BODIPY, except that the signal of exchangeable NH was disappeared, in acid or base solutions. Thus, the acid/base-induced fluorescence switching was achieved. The ON/OFF switch of a fluorescence signal upon addition of acid/base could be cycled back and forth for at least four cycles without significant loss (Fig. S5†).
 | ||
| Fig. 4 (a) Fluorescence spectra of the Z-BODIPY (10 μM) in dichloromethane upon addition of different amounts of trifluoroacetic acid, (b) fluorescence spectra of protonated BODIPY in dichloromethane upon addition of different amounts of DIPEA. λex = 520 nm. | ||
 | ||
| Fig. 5 1H-NMR spectra of (a) Z-BODIPY, (b) protonated BODIPY and (c) protonated BODIPY after adding 3 equiv. of DIPEA (identical as E-BODIPY without the signal of N–H), (d) E-BODIPY in CDCl3. | ||
 | ||
| Scheme 3 Proposed intermediates and transition structure for the rotational isomerization process of Z-BODIPY. | ||
In conclusion, we have successfully developed a multi-stimuli-responsive fluorescent switch by coupling hydrazine with pyridyl-2-aldehyde to BODIPY fluorophore. There were three forms existing: Z-BODIPY, E-BODIPY and protonated BODIPY. Z-BODIPY emitted strong fluorescence, while E-BODIPY and protonated BODIPY were weakly fluorescent. The reversible ON/OFF fluorescence switching among them was realized by irradiation or addition of acid/base. We hope it will inspire researchers to develop multifunctional stimulus-responsive fluorescent materials. This fluorescence switch has potential application in developing new devices for optical data storage and security data encryption.
Acknowledgements
We gratefully acknowledge the financial support from the 973 program (2013CB933800) and the National Natural Science Foundation of China (21402216, 21525206, 21474124), the Fundamental Research Funds for the Central Universities and Beijing Municipal Commission of Education.Notes and references
- Y. Wang, X. Tan, Y.-M. Zhang, S. Zhu, I. Zhang, B. Yu, K. Wang, B. Yang, M. Li, B. Zou and S. X.-A. Zhang, J. Am. Chem. Soc., 2015, 137, 931–939 CrossRef CAS PubMed.
- J. Leblond, H. Gao, A. Petitjean and J.-C. Leroux, J. Am. Chem. Soc., 2010, 132, 8544–8545 CrossRef CAS PubMed.
- Z. Xu, N. J. Singh, J. Lim, J. Pan, H. N. Kim, S. Park, K. S. Kim and J. Yoon, J. Am. Chem. Soc., 2009, 131, 15528–15533 CrossRef CAS PubMed.
- J. Ling, G. Naren, J. Kelly, T. S. Moody and A. P. de Silva, J. Am. Chem. Soc., 2015, 137, 3763–3766 CrossRef CAS PubMed.
- K. Li, Y. Xiang, X. Wang, J. Li, R. Hu, A. Tong and B. Z. Tang, J. Am. Chem. Soc., 2014, 136, 1643–1649 CrossRef CAS PubMed.
- D.-H. Qu, Q.-C. Wang and H. Tian, Angew. Chem., Int. Ed., 2005, 44, 5296–5299 CrossRef CAS PubMed.
- B. Daly, J. Ling and A. P. de Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC.
- S. van de Linde and M. Sauer, Chem. Soc. Rev., 2014, 43, 1076–1087 RSC.
- C. Li, Y. Zhang, J. Hu, J. Cheng and S. Liu, Angew. Chem., Int. Ed., 2010, 49, 5120–5124 CrossRef CAS PubMed.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859–1867 RSC.
- X. Cui, J. Zhao, Y. Zhou, J. Ma and Y. Zhao, J. Am. Chem. Soc., 2014, 136, 9256–9259 CrossRef CAS PubMed.
- T. Fukaminato, T. Doi, N. Tamaoki, K. Okuno, Y. Ishibashi, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 2011, 133, 4984–4990 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC.
- C. Zhao, X. Zhang, K. Li, S. Zhu, Z. Guo, L. Zhang, F. Wang, Q. Fei, S. Luo, P. Shi, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2015, 137, 8490–8498 CrossRef CAS PubMed.
- X.-Z. Wang, Q.-Y. Meng, J.-J. Zhong, X.-W. Gao, T. Lei, L.-M. Zhao, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2015, 51, 11256–11259 RSC.
- F. Wang, L. Zhou, C. Zhao, R. Wang, Q. Fei, S. Luo, Z. Guo, H. Tian and W.-H. Zhu, Chem. Sci., 2015, 6, 2584–2589 RSC.
- O. A. Bozdemir, R. Guliyev, O. Buyukcakir, S. Selcuk, S. Kolemen, G. Gulseren, T. Nalbantoglu, H. Boyaci and E. U. Akkaya, J. Am. Chem. Soc., 2010, 132, 8029–8036 CrossRef CAS PubMed.
- H. Zhu, J. Fan, J. Wang, H. Mu and X. Peng, J. Am. Chem. Soc., 2014, 136, 12820–12823 CrossRef CAS PubMed.
- L.-Y. Niu, Y.-S. Guan, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, J. Am. Chem. Soc., 2012, 134, 18928–18931 CrossRef CAS PubMed.
- L.-Y. Niu, Q.-Q. Yang, H.-R. Zheng, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, RSC Adv., 2015, 5, 3959–3964 RSC.
- L.-Y. Niu, Y.-S. Guan, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Commun., 2013, 49, 1294–1296 RSC.
- T. A. Golovkova, D. V. Kozlov and D. C. Neckers, J. Org. Chem., 2005, 70, 5545–5549 CrossRef CAS PubMed.
- C. Trieflinger, K. Rurack and J. Daub, Angew. Chem., Int. Ed., 2005, 44, 2288–2291 CrossRef CAS PubMed.
- E. Deniz, S. Sortino and F. M. Raymo, J. Phys. Chem. Lett., 2010, 1, 1690–1693 CrossRef CAS.
- L. Kong, H.-L. Wong, A. Y.-Y. Tam, W. H. Lam, L. Wu and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2014, 6, 1550–1562 CAS.
- E. Deniz, S. Ray, M. Tomasulo, S. Impellizzeri, S. Sortino and F. M. Raymo, J. Phys. Chem. A, 2010, 114, 11567–11575 CrossRef CAS PubMed.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- G. Vantomme, S. Jiang and J.-M. Lehn, J. Am. Chem. Soc., 2015, 137, 3138 CrossRef CAS PubMed.
- G. Vantomme, S. Jiang and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 9509–9518 CrossRef CAS PubMed.
- A.-M. Stadler, J. Ramírez and J.-M. Lehn, Chem.–Eur. J., 2010, 16, 5369–5378 CrossRef CAS PubMed.
- L. L. Lao, J.-L. Schmitt and J.-M. Lehn, Chem.–Eur. J., 2010, 16, 4903–4910 CrossRef CAS PubMed.
- H. Qian and I. Aprahamian, Chem. Commun., 2015, 51, 11158–11161 RSC.
- J. T. Foy, D. Ray and I. Aprahamian, Chem. Sci., 2015, 6, 209–213 RSC.
- S. R. Beeren and J. K. M. Sanders, Chem. Sci., 2011, 2, 1560–1567 RSC.
- H. Y. Lee, X. Song, H. Park, M.-H. Baik and D. Lee, J. Am. Chem. Soc., 2010, 132, 12133–12144 CrossRef CAS PubMed.
- H. Li, J. Fan, F. Song, H. Zhu, J. Du, S. Sun and X. Peng, Chem.–Eur. J., 2010, 16, 12349–12356 CrossRef CAS PubMed.
- M. N. Chaur, D. Collado and J.-M. Lehn, Chem.–Eur. J., 2011, 17, 248–258 CrossRef CAS PubMed.
- S. M. Landge, E. Tkatchouk, D. Benítez, D. A. Lanfranchi, M. Elhabiri, W. A. Goddard and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812–9823 CrossRef CAS PubMed.
- X. Su, S. Voskian, R. P. Hughes and I. Aprahamian, Angew. Chem., Int. Ed., 2013, 52, 10734–10739 CrossRef CAS PubMed.
- Y. Yang, X. Su, C. N. Carroll and I. Aprahamian, Chem. Sci., 2012, 3, 610–613 RSC.
- X. Su, M. D. Liptak and I. Aprahamian, Chem. Commun., 2013, 49, 4160–4162 RSC.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2012, 134, 15221–15224 CrossRef CAS PubMed.
- T. F. Robbins, H. Qian, X. Su, R. P. Hughes and I. Aprahamian, Org. Lett., 2013, 15, 2386–2389 CrossRef CAS PubMed.
- T. Rohand, M. Baruah, W. Qin, N. Boens and W. Dehaen, Chem. Commun., 2006, 266–268 RSC.
- O. Dilek and S. Bane, J. Fluoresc., 2011, 21, 347–354 CrossRef CAS PubMed.
- A. D. Mitchell and D. C. Nonhebel, Tetrahedron Lett., 1975, 16, 3859–3862 CrossRef.
- A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2007, 111, 11725–11735 CrossRef CAS PubMed.
- K. K. Kalninsh, J. Struct. Chem., 2008, 49, 427–447 CrossRef CAS.
- D. J. van Dijken, P. Kovaříček, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS PubMed.
- S. M. Landge, E. Tkatchouk, D. Benítez, D. A. Lanfranchi, M. Elhabiri, W. A. Goddard and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812–9823 CrossRef CAS PubMed.
- R. Gawinecki, E. Kolehmainen, H. Janota, R. Kauppinen, M. Nissinen and B. Ośmiałowski, J. Phys. Org. Chem., 2001, 14, 797–803 CrossRef CAS.
- H. Yokoi, S. Hiroto and H. Shinokubo, Org. Lett., 2014, 16, 3004–3007 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01507g |
| This journal is © The Royal Society of Chemistry 2016 |