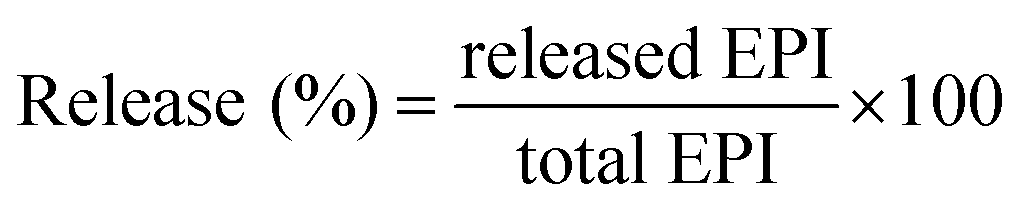DOI:
10.1039/C6RA01482H
(Paper)
RSC Adv., 2016,
6, 26874-26882
Ultrafast synthesis of stabilized gold nanoparticles using aqueous fruit extract of Limonia acidissima L. and conjugated epirubicin: targeted drug delivery for treatment of breast cancer†
Received
18th January 2016
, Accepted 6th March 2016
First published on 8th March 2016
Abstract
In this study, a green chemistry approach was used for the quick synthesis (within 30 seconds) of gold nanoparticles (AuNPs) by using the fruit extract of Limonia acidissima L. The study focused on the formation of L. acidissima L. stabilized AuNPs without using any catalytic agent. The synthesis of the AuNPs was confirmed by observation of the surface plasmon resonance (SPR) band at 537 nm. On the surface of these capped AuNPs, epirubicin (EPI) was conjugated along with activated folic acid (FA) for targeted drug delivery. The EPI–FA–AuNPs complex was characterized using FTIR and a UV-visible spectrophotometer and the AuNPs were characterized by HR-TEM, particle size analyzer, and zeta potential measurements. In vitro stability experiments revealed that the AuNPs were stable in physiological conditions. The in vitro cytotoxicity of free EPI and EPI–FA–AuNPs was investigated against MCF-7 cells, and the results showed that 50% of EPI–FA–AuNPs was enough to achieve inhibition of growth (IC50) and that the amount was lower than that of free EPI. Flow cytometry analysis showed significant reduction in G2/M cells after treatment with EPI–FA–AuNPs, and molecular levels of apoptosis were studied using western blotting. Overall, the results revealed that the EPI–FA–AuNPs have better regression activity on tumor cells than free EPI.
1. Introduction
While chemotherapy drugs are effective, they, unfortunately, have a lot of side effects, namely: (i) conventional cancer drugs are lethal and cause the death of healthy cells as well as cancerous cells; (ii) the drugs have a short lifetime in the body and up to 90% of intravenously delivered drugs may be gathered up by macrophages within the first 5 min of them entering the body; and (iii) the drugs have low solubility, and hence, it is necessary to deliver a larger dose of the conventional anticancer drugs in order to have a good therapeutic index.1 To overcome this problem, we have established a method to synthesize gold nanoparticles (AuNPs) functionalized with folic acid (FA) for targeted delivery of epirubicin (EPI), an anthracycline chemotherapy agent.2 EPI has been clinically used to treat various types of cancer such as breast cancer, lymphomas, sarcomas in soft-tissue, pancreatic cancer, gastric cancer, small-cell lung cancer, and acute leukemia. EPI shows less hematologic or myocardial toxicity at comparable doses.3 AuNPs are identified as favorable candidates for drug delivery applications due to their unique dimensions, tunable surface functionalities, nontoxicity, and controlled drug release capability.4 Green chemistry-based eco-friendly methods are predominantly used for the synthesis of AuNPs instead of chemical synthesis.5 Plant extracts are used as reducing and stabilizing agents to synthesise the nanoparticles. Plant extracts contain different concentrations and combinations of organic reducing agents, which influence the characteristics of the nanoparticles produced.6 Flavonoids, the plant metabolites, contain various functional groups capable of triggering nanoparticle formation. It has been suggested that the tautomeric transformation of flavonoids from the enol-form to the keto-form releases a reactive hydrogen atom that can reduce the metal ions into nanoparticles.7 Fruits of L. acidissima L., contain higher quantities of flavonoids, tyramines, tannins, phytosterols, saponins, glycosides, carbohydrates, vitamins, coumarins, triterpenoids, and amino acids as their chemical constituents.8 The targeted drug delivery system uses the anticancer drug for treatment of cancer cells alone, which reduces the effects of the drug on noncancerous cells and simultaneously increases its efficacy on cancer cells.9 The main aim of using the targeted drug delivery system is to deliver the anticancer drug to the cancerous cells without loss of the drug's efficacy.10,11 The folate receptor (FR) is a meticulously studied ligand for the selective delivery of anticancer drugs on FR-positive tumor cells.12 In general, the FRs are highly up-regulated on the surface of different types of malignant cells.13 In this study, the zebrafish embryo has been used as an in vivo prominent vertebrate model for assessing the toxicity of AuNPs. The zebrafish offers several advantages as a model for in vivo high-throughput drug screening. It is low cost, transparent, and the human genome and the zebrafish genome are highly comparable in terms of tissue types, fertilization, and the development of different systems and their active functions.14 This article deals with the rapid green chemistry-based synthesis of AuNPs that can be used as a carrier for EPI, a drug used to treat breast cancer. Zebrafish embryos were used to examine the developmental toxicity of both normal and drug coupled AuNPs. The efficacy of both EPI and EPI–FA–AuNPs were tested on the MCF-7 cell line (breast cancer).
2. Experimental methods
Material
L. acidissima L. fruit was used for this study. EPI HCl was obtained from SRL Limited, Mumbai (India). Hydrochloroauric acid (HAuCl4), FA, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), dicarboxy aminocarbodiimide (DCC) and 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma-Aldrich Chemicals (USA). MCF-7 cell lines were obtained from National Center for Cell Science (NCCS), Pune, (India). The antibody to actin, Bcl-2, caspases-3 and caspases-8, Fas, FasL, and FADD bax were purchased from Santa Cruz Biotechnology, CA, USA and Neo Markers, USA. Millipore Milli Q water was used for all the experiments. All other chemicals and reagents were of analytical grade.
Activation and attachment of FA to AuNPs
Activation of FA has been reported earlier by Pandey and coworkers.15 It was carried out by dissolving 0.25 g of FA in 20 ml of dimethyl sulfoxide (DMSO), and the mixture was then subjected to sonication for 45 minutes. The carboxylate group of FA was activated by the addition of 0.225 gm of n-hydroxy succinamide (NHS) and 0.125 g of dicarboxy aminocarbodiimide (DDC). The reaction was allowed to take place in an inert environment created by argon gas at 28 °C for 12 hours (FA/NHS/DDC molar ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1). The resultant mixture was filtered through Whatman filter paper and was used for further characterization. Attachment of FA was carried out by adding 9 ml of AuNPs to 1 ml of activated FA. The solution was purged continuously for 4 hours under N2 atmosphere and stirred constantly using a magnetic stirrer. After 4 h, both inlet and outlet valves were sealed, and the solution was allowed to rest for 24 h. After 24 h the compound was filtered using Whatman filter paper and stored at 20 °C. Free FA was removed using a 3000 kDa dialysis bag against phosphate buffered saline (pH 7.2). The post-dialyzed samples were centrifuged at 6000 rpm for 15 min at 20 °C. The pellet was redialyzed with deionized water for 24 h under constant stirring. The samples were subjected to UV-vis spectroscopy at systematic intervals for examination.
:2:1). The resultant mixture was filtered through Whatman filter paper and was used for further characterization. Attachment of FA was carried out by adding 9 ml of AuNPs to 1 ml of activated FA. The solution was purged continuously for 4 hours under N2 atmosphere and stirred constantly using a magnetic stirrer. After 4 h, both inlet and outlet valves were sealed, and the solution was allowed to rest for 24 h. After 24 h the compound was filtered using Whatman filter paper and stored at 20 °C. Free FA was removed using a 3000 kDa dialysis bag against phosphate buffered saline (pH 7.2). The post-dialyzed samples were centrifuged at 6000 rpm for 15 min at 20 °C. The pellet was redialyzed with deionized water for 24 h under constant stirring. The samples were subjected to UV-vis spectroscopy at systematic intervals for examination.
Synthesis of AuNPs–FA–EPI complex
EPI was conjugated to the AuNPs–FA complex for the destruction of cancer cells. Equimolar concentrations of EPI (0.25 mM) and AuNPs–FA (0.25 mM) were subjected to reduction with trimethylamine, 0.5 mM (TEA) using DMSO as a solvent. The molar ratio of AuNPs–FA/EPI/TEA was 1:1:2. The mixture was purged using argon gas with continuous stirring at 50 °C for 4 h. The resultant EPI–FA–AuNPs conjugate was refined using dialysis against nano-pure water for three days with a dialysis tube (MW cut-off of 3000 Da) to remove the excess amount of unbound EPI molecules and DMSO. The water was changed at 6 hour intervals. The entire compound was characterized using UV-vis spectrophotometry and FTIR.
Characterization of AuNPs and EPI–FA–AuNPs
Preliminary characterization of AuNPs and EPI–FA–AuNPs was carried out by FTIR, XRD, HR-TEM, particle size and zeta potential analysis method. LC-MS analysis of the extract was also been carried out. The detailed procedures are given as ESI.†
In vitro stability studies
1 ml of AuNPs was incubated with 0.5 ml each of 0.9% NaCl saline and phosphate buffer saline (PBS, pH 1.2, 4.5, 6.8 and 7.4), respectively, at 37 °C for 48 hours and were analyzed spectrophotometrically by measuring at 480 nm.
In vivo toxicity study of AuNPs in zebrafish embryos
Fertilized eggs of zebrafish were obtained from the natural mating of adult zebrafish, and embryos were collected within 2 hours of spawning. Six healthy embryos from fertilized eggs, approximately 2 hours post fertilization (hpf), were transferred to each well of a 24-well plate containing 1 ml of the E3 medium. The embryos were exposed to different concentrations of AuNPs for 4 days. Duplicates were maintained without the AuNPs as control. Toxicity was assessed by studying the hatching rate, percentage survival rate, and morphology changes in the embryos.
Loading efficiency
From the total concentration of EPI used for loading onto AuNPs, the unloaded drug was removed by separating the supernatant after centrifugation (Eppendorf centrifuge 5430R) at 15000 × g for 15 min. Free EPI present in the supernatant was determined by UV-vis spectrophotometry by measuring at 480 nm.
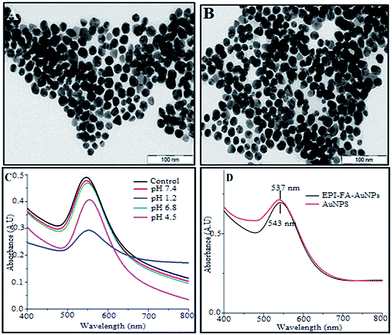
The percentage of drug loading efficiency was calculated using the following formula (1)
| |
 | (1) |
In vitro drug release characteristics
EPI-conjugated AuNPs (equivalent to 1000 μg of EPI) were dialyzed against 100 ml of sodium phosphate buffer (pH 5.7 and 7.4), at 37 °C with continuous stirring at 100 rpm. One milliliter of the sample was withdrawn at specific time intervals and analyzed spectrophotometrically. The sink condition was maintained by replacing equal volumes of the buffer. The release studies were performed in triplicate, and the average was taken. The percentage of drug release was calculated by the using following formula (2):| |
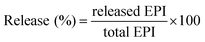 | (2) |
In vitro cytotoxicity studies
Cell viability was measured by MTT assay described by Mossman and coworkers.16 The detailed procedure is given as ESI.†
Apoptosis study
The influence of EPI–FA–AuNPs inducing apoptosis in the breast cancer cells was confirmed using staining methodology.17 The detailed methodology is given as ESI.†
Flow cytometric analysis and western blotting
To investigate the effect of the drug on the cell cycle distribution analysis described by Krishnan,18 western blotting was carried out as described in another paper.19 The detailed methodology descriptions are given in the ESI.†
High content imaging
Localization of EPI–FA–AuNPs and the cytotoxic effect were tested on MCF-7 cell line. Briefly, 2 × 104 cells per well of MCF-7 cancer cell line were seeded on 96 well CellCarrier microplates (PerkinElmer, US). When the cells reached 80% confluence, the medium was changed, and then the cells were treated with AuNPs, EPI, and EPI–FA–AuNPs, and the plate was incubated for 24 hours in a humidified incubator at 37 °C with 5% CO2. The cells were washed twice with ice-cooled PBS, and the drug was localized in live cells using the Operetta High Content Imaging System (PerkinElmer, US).
3. Results and discussion
Characterization of EPI–FA–AuNPs
In the present study, we explain the reducing, stabilizing, and biocompatible properties of L. acidissima L. extract for synthesis of AuNPs. L. acidissima L. has higher quantity of polyphenolic compounds that can actively chelate and reduce metal ions to nanoparticles. It was assumed that the tautomeric conversions of flavonoids from the enol-form to the keto-form may release an activated hydrogen atom that can reduce metal ions to form nanoparticles (yellow color converted to wine red color).
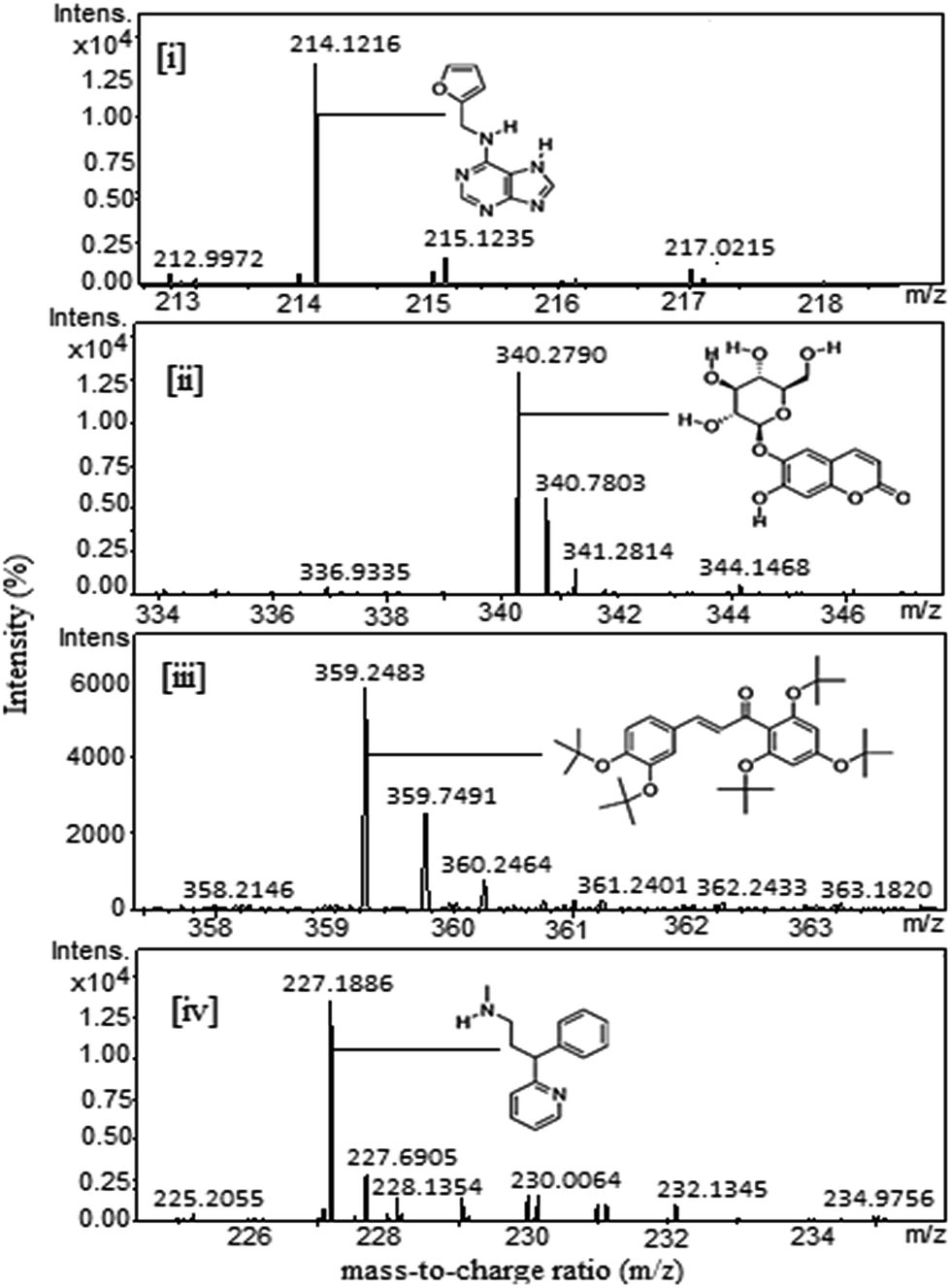
The photochemical profiling of the extract was done by LC-MS analysis which revealed the presence of phytocompounds such as (i) kinetin, (ii) esculin, (iii) 3,4,2′,4′,6′-pentamethoxychalcone, (iv) N-desmethylpheniramine. The identification of compounds was verified by mass fragmentation analysis, and the LC-MS spectra of the compounds are shown in Fig. 1. The combined action of these chemical components and others may be responsible for the observed reduction of the metal ions to form nanoparticles. The AuNPs were formed by simple mixing without applying any external energy (heating, sun light, or microwaving). The resultant mixture produced a significant color change (to wine red) within a few seconds after the addition. Further, our study focused on using the synthesized AuNPs to enable conjugation of biomolecules for different applications in drug delivery (Scheme 1).
 |
| | Fig. 1 LC-ESI-MS/MS analysis of an optimised aqueous extract of Limonia acidissima L. fruit. | |
 |
| | Scheme 1 Schematic representation for the generation of EPI–FA–AuNPs. | |
We observed that the solution containing gold ions (Au3+) and L. acidissima L. turned into wine red (537 nm) within 30 seconds (the synthesis of AuNPs is given in the video file). We found that 75 μL of L. acidissima L. extract Fig. 2A was sufficient to reduce 1 mM HAuCl4. FTIR spectrum of L. acidissima L. extract (Fig. 2B(i)) shows a peak at 3448 cm−1, which could be attributed to the phenolic hydroxyls (O–H bond) in extracts, the absorption peaks at 1048 and 1612 cm−1 representing costretching functional present in the extract. Further, carboxyl stretching vibration peak formed at 1730 cm−1.
 |
| | Fig. 2 (A) Optimization of AuNPs by changing various concentrations of (L. acidissima L. extract) while keeping the gold solution constant. (B) FT-IR study of: (i) L. acidissima L. extract (ii) AuNPs (iii) FA (iv) EPI (v) EPI–FA–AuNPs. | |
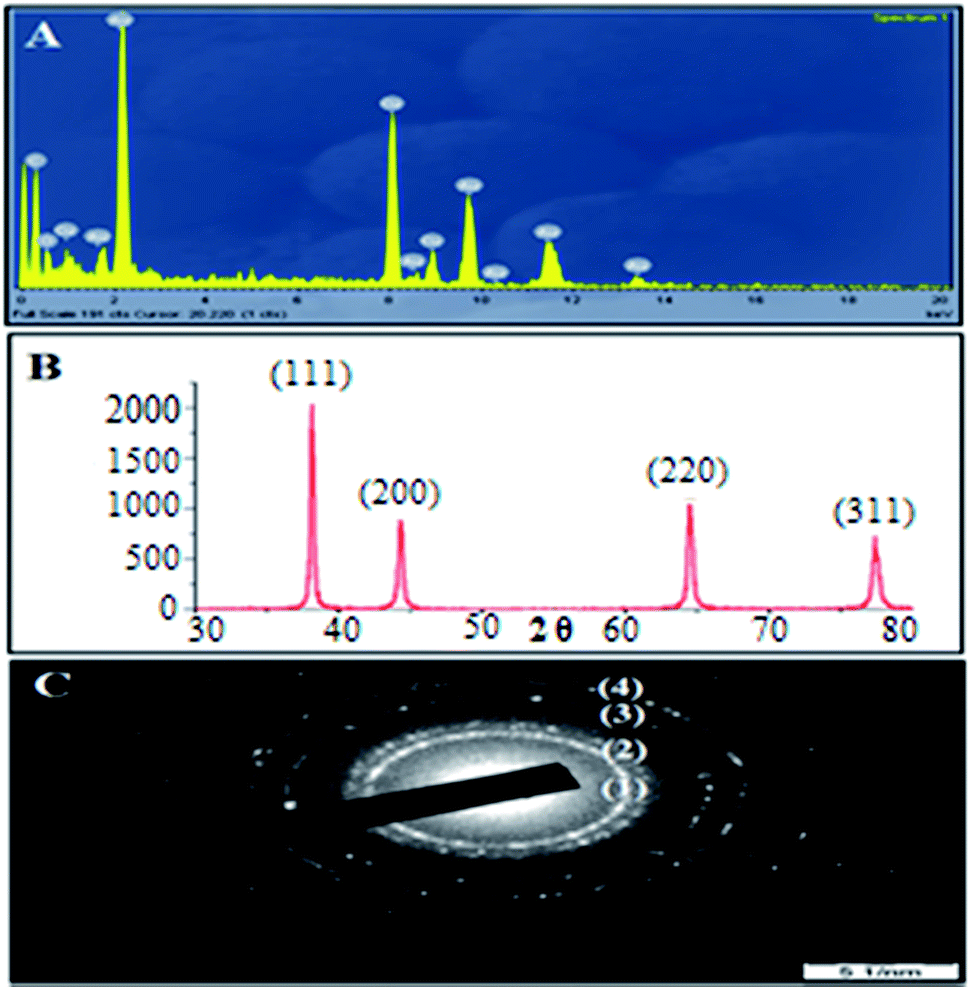
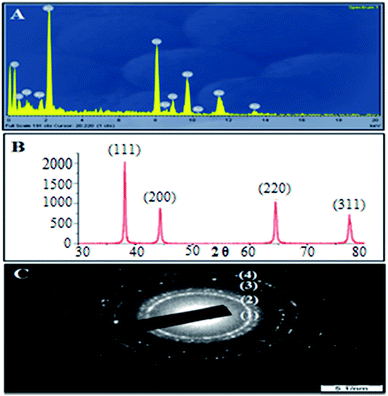
However, in the FTIR spectrum of AuNPs Fig. 2B(ii), the peak at 3420 cm−1 becomes comparatively narrow, which could be attributed to the phenolic hydroxyls (O–H bond) confirming that the phenolic hydroxyls react with the AuNPs resulting in the partial destruction of the hydrogen bonds in the molecules of the L. acidissima L. extract. A shift in the peak from 1048 cm−1 to 1017 cm−1 also indicates that L. acidissima L. extract interacts with AuNPs through its adjacent phenolic hydroxyls and/or formed quinones. The comparison of FTIR spectra of bare AuNPs, FA, EPI, and EPI–FA–AuNPs shows that the spectrum of FA–AuNPs exhibits the characteristic IR absorption peaks of FA Fig. 2B(iii), showing a peak at 1697 cm−1 (amide I stretching), 1603 cm−1 (amide II stretching), and 1481 cm−1 (hetero-ring, conjugated double bond). FTIR spectra of EPI and EPI–FA–AuNPs are shown in Fig. 2B(iv and v). FTIR spectra of EPI clearly confirm the basic structural units such as OH, CH, and carbonyl groups carboxyl groups by the peaks that appear at 3300 cm−1, 2940 cm−1, and 1620 cm−1, respectively. The FTIR spectrum of EPI–FA–AuNPs shows the peaks at 3300 cm−1 and 2940 cm−1, demonstrating the OH and CH content of EPI–FA–AuNPs. Moreover, the disappearance of the peak at 1730 cm−1 describes the amide bond formation between the carboxyl functional of EPI and amine functional of FA. These spectra confirm the formation of EPI–FA–AuNPs. Energy dispersive X-ray (EDX) spectrum analysis of AuNPs showed the presence of AuNPs in the sample. In Fig. 3A reveals a strong and typical optical absorption peak at approximately 2.2 keV, which could be attributed to the SPR of the metallic Au nanocrystals. The crystalline structure of nanoparticles was determined based on X-ray diffraction (XRD) analysis. The crystalline peaks that were identified as AuNPs based on XRD analysis showed that the intense peaks of reflected radiation (Bragg peaks) were at the points (1 1 1), (2 0 0), (2 2 0), and (3 1 1) and were the diffraction lines of the face-centered-cubic (fcc) gold are shown in Fig. 3B. The selected area of electron diffraction (SAED) outline of the AuNPs showing that the rings designated 1, 2, 3, and 4 arise because of the reflections from (1 1 1), (2 0 0), (2 2 0), and (3 1 1) are shown in Fig. 3C. The morphological results (HRTEM) images Fig. 4A reveals that the AuNPs and EPI–FA–AuNPs appear to be nearly spherical in shape as seen in Fig. 4B. The stability of EPI–FA–AuNPs in different buffers was examined in Fig. 4C, and only slight changes (<15%) were observed in the SPR band at 537 nm when the AuNPs were exposed to different buffers for 48 hours. However, at pH 1.2, about 20% decline in the absorbance at 537 nm was observed compared with the absorbance of EPI–FA–AuNPs at pH (7.4). The observed reduction in the absorption of EPI–FA–AuNPs in extremely acidic conditions could be attributed to the limited aggregation of EPI–FA–AuNPs as a result of the screening of the negative charge on the external of the AuNPs at pH (1.2). The results from these in vitro stability studies established that the AuNPs were intact and demonstrated excellent in vitro stability in biological fluids at various pH.20 EPI binding onto the AuNPs was further confirmed by the shift of the SPR band in Fig. 4D towards a higher wavelength (from 537 nm to 543 nm). The percentage of EPI loaded onto the AuNPs was determined based on EPI content in the obtained pellet, and it was found that 52 ± 4% of the drug could be loaded. Inductively coupled plasma optical emission spectrophotometer (ICP-OES) was performed to quantify the amount of gold in the aqueous solutions of AuNPs, and the same was used in further studies.
 |
| | Fig. 3 (A) The EDAX spectrum, (B) X-ray diffraction (XRD) spectrum, (C) SAED pattern of L. acidissima L. fruit extract reduced AuNPs. | |
 |
| | Fig. 4 HRTEM image of (A) AuNPs (B) EPI–FA–AuNPs, (C) the UV-vis spectra showing the in vitro stability of EPI–FA–AuNPs in the pH range 1.2–7.4 and PBS, (D) AuNPs conjugated EPI–FA–AuNPs. | |
Particle size and zeta potential measurements
The hydrodynamic particle size of the AuNPs was found to be 134 ± 4 nm with a polydispersity index of 0.219 are shown in Fig. 5(i). The average hydrodynamic diameter of EPI–FA–AuNPs is 139 ± 3 nm with a polydispersity index of 0.249 shown in Fig. 5(ii). A low polydispersity index shows that the particle size distribution of the AuNPs and EPI–FA–AuNPs is uniform. Zeta potential is considered an essential parameter to study the surface charge of the nanoparticle surface and predict the long-term stability of the nanoparticles.21 The zeta potential of the AuNPs was found to be −27.9 mV Fig. 5(iii) indicating that the AuNPs were properly capped with anionic (carboxyl group) FA. EPI–FA–AuNPs were isolated by centrifugation and then suspended in an aqueous solution to quantify the electrophoretic mobility. The calculated zeta potential of EPI–FA–AuNPs was −16.0 mV as seen in Fig. 5(iv). Though the surface charge is reduced to some extent, EPI–FA–AuNPs has found to be stable without aggregation when analysed after six-month storage period. Hence, it is clear that EPI–FA–AuNPs are highly stable over a reasonable period of time.
 |
| | Fig. 5 (i) The particle size distribution of AuNPs (ii) EPI–FA–AuNPs (iii) the zeta potential distribution of AuNPs in an aqueous medium. (iv) The zeta potential of distribution EPI–FA–AuNPs in an aqueous medium. | |
Toxicity of AuNPs in zebrafish embryos
Zebrafish embryos, which are ideal organisms, were used to investigate the developmental toxicity of AuNPs without EPI–FA.22 The survival rate was better for the embryos that had hatched within 96 hours post fertilization, as compared with the control group as seen in Fig. 6A(i). The mortality level was indicated by the dead embryos 96 hours post fertilization as compared to the control group in Fig. 6A(ii). The survival rate and mortality rate examined shows no apparent toxicity at various concentrations ranging from 50 to 450 μg mL−1.23 The AuNPs-treated embryos were observed using a compound stereo microscope to find abnormalities such as pericardial edema, pigmentation, deformity of the pericardial sac of the larvae, and deformity of the tail. Exposure of zebrafish egg embryos to AuNPs did not produce any seeming toxicity in the development of the zebrafish are shown in Fig. 6A(iii). Hence, this investigation of results clearly indicated that the AuNPs might be carriers for drug delivery applications.
 |
| | Fig. 6 (A) (i) Graph representing the toxicity of AuNPs in terms of survival rate (%) of larvae, (ii) mortality rate (%) analysis of AuNPs toxicity, (iii) examples of zebrafish larvae exposed to control and various concentrations of AuNPs. Significant differences compared to control calculated with the one-way ANOVA test and are indicated by ***p < 0.001. (B) In vitro release profiles of EPI from the EPI–FA–AuNPs and free EPI in phosphate buffer solution (pH 5.7 and 7.4) at 37 °C. | |
Release profile of EPI–FA–AuNPs
The in vitro study of pH-controlled EPI release from the EPI–FA–AuNPs was performed in PBS (pH = 7.4) and acetate buffer (pH = 5.7) at 37 °C, pH (7.4) and pH (5.7) being near the physiological and endosomal pH conditions, respectively, of a cancer cell. The results are presented in Fig. 6B. The pH-dependent drug release is also considered to be an essential factor in cancer therapy.24 The EPI–FA–AuNPs displays the slow and controlled release of the drug, with the release rate calculated showing negligible release when compared to that of free EPI in acidic and neutral atmospheres, respectively.
Furthermore, the release efficiency of the drug from the EPI–FA–AuNPs was rapid, higher at pH (5.7) than at pH (7.4) EPI from EPI–FA–AuNPs in pH (7.4) will help to reduce toxicity of EPI to the normal tissue since the physiological pH of the body is maintained at pH (7.4).25
In vitro cytotoxicity studies
To determine the cytotoxic effect of EPI and EPI–FA–AuNPs, cell viability study was done by a standard MTT-reduction assay with slight modifications.26 The results of the MTT assay for free and EPI–FA–AuNPs on MCF-7 cell lines are shown in Fig. 7A. The IC50 value for EPI–FA–AuNPs was found to be around 2 μg mL−1, while that for free EPI ranged from 28 μg mL−1 on the MCF-7 cells. These results show that the EPI–FA–AuNPs exhibited better cytotoxic activity than free EPI. This could possibly be due to the result of the variations in the cellular uptake profile leading to better activity of EPI–FA–AuNPs as suggested by Ormrod and coworkers.27 The results of this study suggests that FA functionalized AuNPs can effectively deliver the drug to MCF-7 cancer cells by means of active targeting.
 |
| | Fig. 7 (A) In vitro cytotoxicity studies of drug EPI and EPI–FA–AuNPs in MCF-7 cell lines. All assays were performed in triplicate, and the mean ± standard deviations are shown. (B) Images of MCF-7 cell lines visualized under inverted microscope. (i) Control, (ii) blank, (iii) EPI, and (iv) EPI–FA–AuNPs. | |
Apoptosis study
Apoptotic changes, cytotoxic effect caused by EPI–FA–AuNPs occurred in MCF-7 cells when treated using acridine orange/ethidium bromide differential staining method. The stained cells characterized to EPI–FA–AuNPs caused more effective cell death than free EPI. The number of nonviable cells increased dramatically after treatment with EPI–FA–AuNPs in MCF-7 cells are shown in Fig. 7B(iv). The red color is due to the nonviable cells (dead cells), and it was revealed that EPI–FA–AuNPs treatment resulted in a significant increase in apoptosis compared to EPI treatment. The control and blank cells in Fig. 7B(i and ii), the condensed nuclei, as well as the membrane blebbing fluoresced uniformly bright green indicating (early apoptotic) that they did not undergo any apoptotic changes.
Effect of EPI–FA–AuNPs on cell cycle analysis
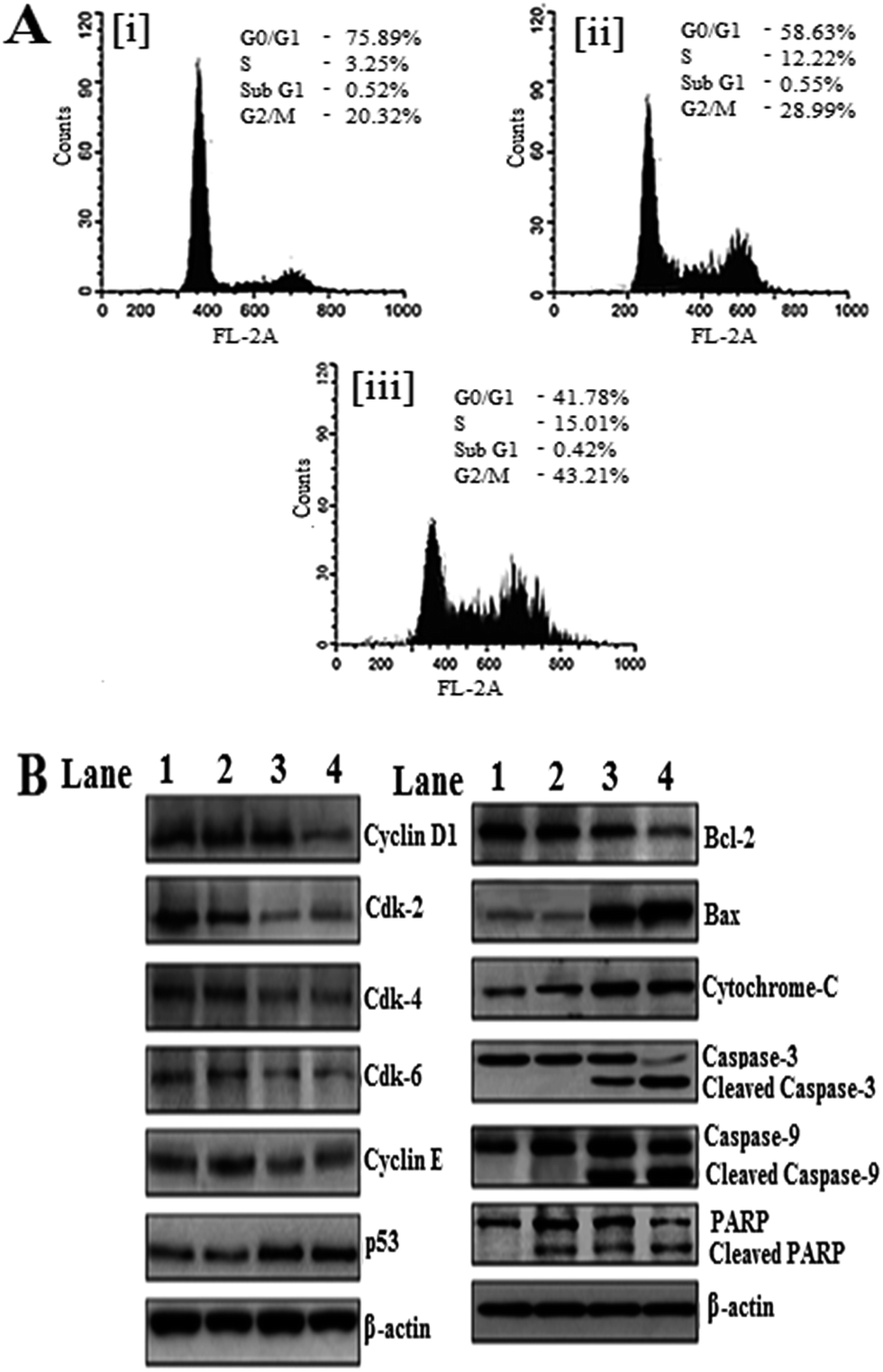
Generally, the flow cytometry study was performed to investigate whether apoptosis and the cell cycle arrest are closely related. In our study, the effect of EPI and EPI–FA–AuNPs-on cell cycle (MCF-7 cells) was studied. Fig. 8A shows the cell cycle analysis of (i) control, (ii) EPI and (iii) EPI–FA–AuNPs. Control cells after 36 hours had 20.3% the proportion of cells in the G2/M phase in Fig. 8A(i), and the EPI and EPI–FA–AuNPs produced a dramatic increase of 28.95% in Fig. 8A(ii) and 43.21% are shown in Fig. 8A(iii) in the G2/M population, respectively. These data clearly prove that the cell cycle is arrested significantly at the G2/M phase in EPI–FA–AuNPs induced MCF-7 cells.28 The anti-cancer therapy completely eradicates the cancer cells by triggering different caspase-mediated cell death pathways,29 which confirms the activation of the apoptotic pathway after EPI–FA–AuNPs' exposure.
 |
| | Fig. 8 (A) Effects of EPI–FA–AuNPs on cell cycle analysis. MCF-7 non-small cell breast cancer cells were cultured for 36 hours (i) control (ii) EPI (iii) EPI–FA–AuNPs. (B) Western blot, illustrating the different protein expressions in apoptosis after treatment with EPI and EPI–FA–AuNPs. Lane 1: control, lane 2: AuNPs, lane 3: EPI, lane 4: EPI–FA–AuNPs. | |
Western blot analysis
The western blot results showed mechanisms of EPI–FA–AuNPs-mediated cell death; the apoptotic regulators have been measured using mRNA and protein expression patterns. Cyclin D1 regulates the proliferation process by adjusting its expression levels and, accordingly, regulates the cell cycle control machinery.30 EPI controls the reduction of cyclin D1 levels in tumors.31 The results as seen in Fig. 8B showed that EPI–FA–AuNPs inhibited the function of cyclin D1, and its expression was dramatically diminished. Cyclin E showed a high intense band when AuNPs and native EPI-treated cells were compared. The functions of cyclin-dependent kinase CDK4, CDK6, and CDK2 mediated phosphorylation of the retinoblastoma (Rb) family of tumor suppressor proteins during apoptosis were also observed.32 EPI–FA–AuNPs induced apoptosis in cancer cells was evident from the reduced expression of anti-apoptotic CDK6, CDK4, and CDK2 in MCF-7 cells. In this study on the efficacy of CDK inhibition as a means to arrest the proliferation of HCC, β-actin was used as a loading control, and it showed similar expression in all the lanes. Intrinsic pathways of western blot studies are shown in Fig. 8B. The present study also investigated EPI–FA–AuNPs induced up-regulation of Bax proteins and down-regulation of Bcl-2 proteins in EPI–FA–AuNPs cells. p53 is a leading switch that coordinates stress signals between apoptosis and cell cycle arrest33 is shown in Fig. 8B. EPI–FA–AuNPs induced increase in the levels of p53, the tumor suppressor protein. Caspases, a family of cysteine acid proteases, can be regarded as key factors in apoptosis.34 In Fig. 8B shows that EPI–FA–AuNPs treated cells induced cleavage of caspase 3 and caspase 9. On the whole, these pH results clearly indicate that the molecular level apoptosis was induced when MCF-7 cells were treated with EPI–FA–AuNPs. Cell death was further studied based on PARP cleavage after treatment with EPI–FA–AuNPs, as seen in Fig. 8B. Our studies showed that PARP proteins cleaved into fragments after treatment with EPI–FA–AuNPs.
High content imaging
The cellular uptake study was made in MCF-7 cell line for EPI, AuNPs, and EPI–FA–AuNPs using high content imaging methods, and the results are shown in Fig. 9. EPI–FA–AuNPs' localization into the cell was compared with EPI in which bright field and fluorescent images were taken. The merged images help to calculate the localization of EPI–FA–AuNPs. Control cells without the exposure to EPI and AuNPs indicated no fluorescence.35 The EPI–FA–AuNPs-treated cells showed maximum localization of EPI–FA–AuNPs into cells compared to EPI-treated cells.
 |
| | Fig. 9 Drug localization studies of control, EPI, AuNPs, and EPI–FA–NPs in MCF-7 cell line. | |
4. Conclusion
To summarize the present study, a rapid and green chemistry-based method, which relies on the reduction of gold ions by L. acidissima L. extract acting as both reducing and capping agents, has been established to synthesize AuNPs. The EPI–FA–AuNPs exhibits prominent anticancer activity against MCF-7 cells. Results of the toxicity study in the zebrafish embryo model revealed that no significant malformation occurred, ensuring that the AuNPs nanoparticles are highly compatible for drug-delivery applications. Furthermore, maximum localization into cells compared with EPI in the MCF-7 cell line was observed. The EPI–FA–AuNPs could be promising for targeting breast cancer with the enhanced therapeutic activity of EPI.
Acknowledgements
The authors are thankful to the University Grants Commission (UGC), India for financial support through the project grant no. F1-17.1/2011-12/RGNF-SC-TAM-2228/(SA-III) to one of the authors, C. Senthil Kumar. We thank National Facility for Drug Development for Academia, Pharmaceutical and Allied Industries (NFDD) for particle size analyser facility LC-MS and FTIR studies.
References
- B. Asadishad, M. Vossoughi and I. Alemzadeh, Ind. Eng. Chem. Res., 2010, 49, 1958–1963 CrossRef CAS.
- W. Tian, X. Ying, J. Du, J. Guo, Y. Men, Y. Zhang, R. Li, H. Yao, J. Lou, L. Zhang and W. L Lu, Eur. J. Pharmacol., 2010, 41, 232–243 CrossRef CAS PubMed.
- C. G. Kumar, Y. Poornachandra and S. K. Mamidyala, Colloids Surf., B, 2014, 123, 311–317 CrossRef PubMed.
- P. Joshi, S. Chakraborti, J. E. Ramirez-Vick, Z. A. Ansari, V. Shanker, P. Chakrabarti and S. P. Singh, Colloids Surf., B, 2012, 95, 195–200 CrossRef CAS PubMed.
- P. R. Devi, C. S. Kumar, P. Selvamani, N. Subramanian and K. Ruckmani, Mater. Lett., 2015, 139, 241–244 CrossRef.
- A. K. Mittal, Y. Chisti and U. C. Banerjee, Biotechnol. Adv., 2013, 31, 346–356 CrossRef CAS PubMed.
- V. V. Makarov, A. J. Love, O. V. Sinitsyna, S. S. Makarova, I. V. Yaminsky, M. E. Taliansky and N. O. Kalinina, Acta Naturae, 2014, 6, 20 Search PubMed.
- D. T. Priyadarsini, V. Maheshu, M. Vishnupriya, S. Nishaa and J. M. Sasikumar, Free Radicals Antioxid., 2013, 3, S62–eS69 CAS.
- T. Betancourt, B. Brown and L. Brannon-Peppas, Nanomedicine, 2007, 2, 219–232 CrossRef CAS PubMed.
- K. K. Jain, Technol. Cancer Res. Treat., 2005, 4, 311–313 CrossRef CAS PubMed.
- D. C. Li, X. K. Zhong and Z. P. Zeng, J. Controlled Release, 2009, 138, 103–112 CrossRef CAS PubMed.
- Y. Lu and P. S. Low, Adv. Drug Delivery Rev., 2012, 64, 342–352 CrossRef.
- Z. Zhang, J. Jia, Y. Lai, Y. Ma, J. Weng and L. Sun, Bioorg. Med. Chem., 2010, 18, 5528–5534 CrossRef CAS PubMed.
- M. M. Poggi, D. N. Danforth, L. C. Sciuto, S. L. Smith, S. M. Steinberg, D. J. Liewehr, C. Menard, M. E. Lippman, A. S. Lichter and R. M. Altemus, Cancer, 2003, 98, 697–702 CrossRef PubMed.
- S. Pandey, G. Oza, A. Mewada, R. Shah, M. C. Thakur and M. Sharon, J. Mater. Chem. B, 2013, 1, 1361 RSC.
- T. Mossman, J. Immunol. Methods, 1983, 65, 55–63 CrossRef.
- S. Karthik, R. Sankar, K. Varunkumar and V. Ravikumar, Biomed. Pharmacother., 2014, 68, 327–334 CrossRef CAS PubMed.
- A. Krishnan, J. Cell Biol., 1975, 66, 188–193 CrossRef.
- L. C. Tu, C. K. Chou, C. Y. Chen, Y. T. Chang, Y. C. Shen and S. F. Yeh, Biochim. Biophys. Acta, 2004, 1672, 148–156 CrossRef CAS PubMed.
- M. Ganeshkumar, M. Sathishkumar, T. Ponrasu, M. G. Dinesh and L. Sugun, Colloids Surf., B, 2013, 106, 208–216 CrossRef CAS PubMed.
- D. Pooja, S. Panyaram, H. Kulhari, B. Reddy, S. S. Rachamalla and R. Sistla, Int. J. Biol. Macromol., 2015, 80, 48–56 CrossRef CAS PubMed.
- P. Sharma, S. Sharma, V. Patial, D. Singh and Y. S. Padwad, Clinical Queries: Nephrology, 2014, 3, 97–105 CrossRef.
- J. A. Harris, A. G. Cheng, L. L. Cunningham, G. MacDonald, D. W. Raible and E. W. Rubel, Journal of the Association for Research in Otolaryngology, 2003, 4, 219–234 CrossRef PubMed.
- C. S. Kumar, M. D. Raja, D. S. Sundar, M. Gover Antoniraj and K. Ruckmani, Carbohydr. Polym., 2015, 128, 63–74 CrossRef CAS PubMed.
- E. Rafiee and S. Eavani, Mater. Sci. Eng., C, 2014, 39, 340–343 CrossRef CAS PubMed.
- M. Tariq, M. D. Aftab Alamb, A. T. Singhc, Z. Iqbala, A. K. Pandab and S. Talegaonkara, Colloids Surf., B, 2015, 128, 448–456 CrossRef CAS PubMed.
- D. Ormrod, K. Holm, K. Goa and C. Spencer, Drugs Aging, 1999, 15, 389 CAS.
- C. M. Halloran, P. Ghaneh, S. Shore, W. Greenhalf, L. Zumstein, D. Wilson, J. P. Neoptolemos and E. Costello, Eur. J. Gen. Med., 2004, 6, 514–525 CrossRef CAS PubMed.
- S. Ghavami, M. Hashemi, S. R. Ande, B. Yeganeh, W. Xiao, M. Eshraghi, C. J. Bus, K. Kadkhoda, E. Wiechec, A. J. Halayko and M. Los, J. Med. Genet., 2009, 46, 497–510 CrossRef CAS PubMed.
- G. PankajRoy, Cyclin D1 and breast cancer, Breast, 2006, 15, 718–727 CrossRef PubMed.
- M. A. V. Velázquez, Z. Li, M. Casimiro, E. Loro, N. Homsi and R. G. Pestell, Future Oncol., 2011, 7, 753–765 CrossRef PubMed.
- C. G. Murphy and M. N. Dickler, Oncologist, 2015, 20, 483–490 CrossRef CAS PubMed.
- I. A. Hedenfalk, B. Baldetorp, A. Borg and S. M. Oredsson, Cytometry, 1997, 29, 321–327 CrossRef CAS PubMed.
- W. L. Sun, J. Chen, Y. P. Wang and H. Zheng, Autophagy, 2011, 7, 1035–1044 CrossRef CAS PubMed.
- G. Yordanov, R. Skrobanska and A. Evangelatov, Colloids Surf., B, 2012, 92, 98–105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01482h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.