
Synthesis of wrinkled graphene hybrids for enhanced visible-light photocatalytic activities†
Abstract
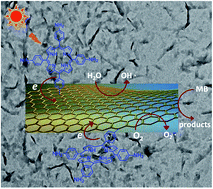
Wrinkled graphene hybrids covalently modified with porphyrin were controllably synthesized and confirmed using Fourier transform infrared spectroscopy, UV-vis spectroscopy, fluorescence emission spectroscopy, thermogravimetric analysis, Raman spectroscopy, X-ray photoelectron spectroscopy, and transmission electron microscopy. Compared with the planar graphene hybrids covalently modified with porphyrin, the wrinkled graphene hybrids exhibit enhanced photocatalytic activity in the degradation of methylene blue under visible light. The cause of the formation of wrinkled graphene hybrids was analyzed and ascribed to the porphyrin interaction on the basal planes of graphene. This investigation might not only provide a new pathway toward the controllable synthesis of graphene hybrid materials with wrinkled morphology, but also a new approach to improving the photocatalytic performance of organic dye-sensitized graphene hybrid materials.
 Please wait while we load your content...
Please wait while we load your content...

