
Asymmetric immobilization of antibodies on a piezo-resistive micro-cantilever surface†
Abstract
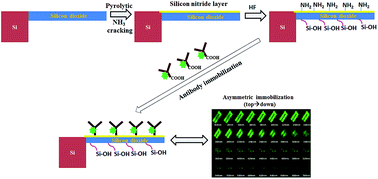
For cantilever-based MEMS sensors, selective chemical modification of the sensing surface is used for the detection of chemical and biological analytes. One of the key challenges in these kind of applications is to obtain an increased level of sensitivity, which largely depends on the surface coverage of the respective probe molecules on one of the cantilever surfaces (asymmetric immobilization). Usually, a blocking layer of another material like gold is deposited on one side of the cantilever surface to obtain layers with different chemical properties. In this paper, we report a novel approach of grafting antibodies on a single side of micro-fabricated piezo-resistive cantilevers. The microcantilevers were fabricated with polysilicon as the piezo-resistive layer, sandwiched between silicon dioxide structural layers. In order to immobilize antibodies on one of the cantilever surfaces (top or bottom), a thin layer of amorphous silicon was deposited on the surface where immobilization needs to be done. This amorphous silicon layer was partially modified into silicon nitride by a Hot-Wire Chemical Vapour Deposition (HWCVD) mediated pyrolytic ammonia-cracking process. The developed selective surface modification protocol for nitride and silicon dioxide surface was studied using X-ray photoelectron spectroscopy (XPS), ellipsometry and contact-angle goniometry. The protocol was first developed on silicon wafer surface, further extending its application onto the piezo-resistive micro-cantilevers. Consequentially, asymmetric immobilization was successfully carried out by selectively grafting antibodies onto the top (modified into nitride) surface of the micro-cantilever by passivating the bottom oxide surface. It was verified using fluorescence microscopy and laser confocal microscopy. Further, we have demonstrated that asymmetrically modified cantilevers show an increased electrical response (20–100%) in comparison to symmetrically modified ones.
 Please wait while we load your content...
Please wait while we load your content...

