
Impact of the functional group in the polyanion of single lithium-ion conducting polymer electrolytes on the stability of lithium metal electrodes†
Abstract
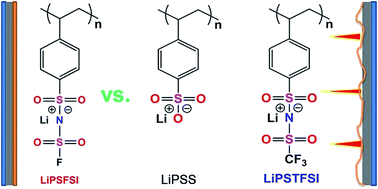
A novel single lithium-ion (Li-ion) conducting polymer electrolyte composed of lithium poly[(4-styrenesulfonyl)(fluorosulfonyl)imide] (LiPSFSI) and poly(ethylene oxide) (PEO) exhibits a high Li-ion transference number (tLi+ = 0.90) and sufficient electrochemical stability for use in Li batteries. The ionic conductivities of the LiPSFSI/PEO blended polymer electrolytes are higher than those of the lithium poly(4-styrenesulfonate) (LiPSS)/PEO electrolyte and are comparable to those of the lithium poly[(4-styrenesulfonyl)(trifluoromethanesulfonyl)imide] (LiPSTFSI)/PEO electrolyte in the temperature range of 25–90 °C. More importantly, the complex of LiPSFSI/PEO exhibits excellent interfacial compatibility with the Li metal electrode compared to both those of the LiPSS/PEO and LiPSTFSI/PEO electrolytes.
 Please wait while we load your content...
Please wait while we load your content...

