
Aryl-triazolyl peptides for efficient phase selective gelation and easy removal of dyes from water†
Bhartendu K. Srivastava and
Muraleedharan K. Manheri*
Department of Chemistry, Indian Institute of Technology Madras, Chennai, 600036, India. E-mail: mkm@iitm.ac.in; Fax: +91 44 2257 4202; Tel: +91 44 2257 4233
First published on 15th March 2016
Abstract
Fine-tuning the gelation ability of aryl triazolyl peptide 1 by C-terminal modification led to the identification of 2 with the remarkable ability to form highly transparent gels in a wide range of solvents including oils. Good rheological properties, effective phase-selective gelation in short time periods, and usefulness in dye removal from water are the other highlights.
Low molecular weight organogelators (LMWOGs) are at the forefront of research due to their direct application in areas like drug delivery, soft optical devices and oil spill recovery.1 Although from different structural classes like amino acids/peptides,2 sugars,3 aromatics or their amphiphilic derivatives,4 they have the unique ability of forming fibrillar aggregates through self-assembly; these primary structures further develop into entangled three-dimensional networks capable of holding large amounts of solvent – of the order of 104 solvent molecules per molecule of the gelator, in the case of many supergelators.5 Naturally, hydrogen bonding, π-stacking, electrostatic interactions, hydrophobic effects etc. form the basis of these super-structures. The nature, directionality, or availability of chemical groups associated with these interactions dictate the gel strength and solvent preferences.
Despite having large number of examples, our ability to rationally design a gelator with predefined properties is minimal, and serendipity seems to be the origin of most of the systems known thus far. Response to stimuli such as temperature, light, pH, metal ions etc.6 or the ability to bring phase-selective gelation7 have been major focus of studies involving this class of compounds. Identification of organic compounds which offer opportunity to access a wide window of gelation through introduction of solvent-specific groups at appropriate locations will be greatly advantageous. This report introduces the aryl-triazolyl peptide 1 (Fig. 1) as a new low-molecular weight organogelator with provision for broadening the gelation window through C-terminal modification (e.g. 2).
 | ||
| Fig. 1 Structures of aryl-triazolyl peptide-based organogelators. | ||
It was synthesized in a straightforward manner using anthranilic acid as the starting material (ESI, Scheme S1 and S2†). This class of peptides were originally designed as part of our on-going studies on oligomers with specific folding-, aggregation- and metal-binding propensities.8 Extraordinary ability of 1 to gelate aromatic solvents like benzene, toluene and mesitylene with CGC values of 1.5, 0.6, 0.1% w/v respectively came as a pleasant surprise. This encouraged us to carry out more focussed studies to understand the structural characteristics responsible for solvent entrapment. The other solvent in which 1 showed excellent gelation was CCl4 (0.08% w/v). While insolubility in hexane and oils (diesel, petrol, kerosene etc.) precluded gelation in these solvents, good solvent entrapment was seen in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 EtOAc–hexane mixture. Likely due to disruption of fibrillar networks through solvation, gel formation was inefficient in MeOH and EtOH. However, on increasing the hydrocarbon content, as in isopropanol and dodecanol, the supramolecular network of 1 was sufficiently stable to give strong gels (ESI, Table S1†). Thus, aromatic solvents, CCl4, isopropanol and dodecanol seemed to give a balanced solubility for 1 and favour the self-assembly essential for gelation. It is reasonable to assume that proper conformational bias and aggregation of 1 under these conditions happen in such a way as to give adequate π-stacking/H-bonding possibilities along the solvent-exposed surface of the supramolecular network. As the terminals are likely to be more solvent exposed, we prepared the derivatives 2 and 3 possessing a dodecyl chain at C- and N-terminus respectively. Of these, 2 not only gelated aromatic solvents, but was also efficient in immobilizing hydrocarbons like heptane–THF (4:1), diesel, petrol, vegetable oils etc (ESI, Table S1†). These gels were transparent and had low CGC values (% w/v); for petrol and diesel, it was 0.1 and 0.3 respectively. In comparison, 3 with C12-chain at the N-terminus was poor in gelating aromatic as well as aliphatic solvents. Since conformational bias with proper distribution of donor acceptor- and π-stacking moieties for self-assembly is essential for gelation, the C-terminal region seems to tolerate chemical modification to a larger extent without compromising the properties. Obviously, the self-assembly profiles of 2 and 3 are going to be different which also should be responsible for the difference in the gelation ability.
:1 EtOAc–hexane mixture. Likely due to disruption of fibrillar networks through solvation, gel formation was inefficient in MeOH and EtOH. However, on increasing the hydrocarbon content, as in isopropanol and dodecanol, the supramolecular network of 1 was sufficiently stable to give strong gels (ESI, Table S1†). Thus, aromatic solvents, CCl4, isopropanol and dodecanol seemed to give a balanced solubility for 1 and favour the self-assembly essential for gelation. It is reasonable to assume that proper conformational bias and aggregation of 1 under these conditions happen in such a way as to give adequate π-stacking/H-bonding possibilities along the solvent-exposed surface of the supramolecular network. As the terminals are likely to be more solvent exposed, we prepared the derivatives 2 and 3 possessing a dodecyl chain at C- and N-terminus respectively. Of these, 2 not only gelated aromatic solvents, but was also efficient in immobilizing hydrocarbons like heptane–THF (4:1), diesel, petrol, vegetable oils etc (ESI, Table S1†). These gels were transparent and had low CGC values (% w/v); for petrol and diesel, it was 0.1 and 0.3 respectively. In comparison, 3 with C12-chain at the N-terminus was poor in gelating aromatic as well as aliphatic solvents. Since conformational bias with proper distribution of donor acceptor- and π-stacking moieties for self-assembly is essential for gelation, the C-terminal region seems to tolerate chemical modification to a larger extent without compromising the properties. Obviously, the self-assembly profiles of 2 and 3 are going to be different which also should be responsible for the difference in the gelation ability.
Since gelators of diesel and petrol can find application in oil-spill recovery,9 we studied phase-selective gelation of 2 with these fuels. For a LMWOG to be useful in ‘real-life’ situations, the PSG process should be fast and efficient. ‘Instant gelation’ of diesel by N-terminal modified phenyl glycine reported by Basak et al.7d and similar results from N-acetylglucosamine-based gelator from the laboratory of Mukherjee et al.7c are some important developments in this area. Conventional heating–cooling technique is not suitable for practical applications and a biphasic mixture of organic solvent and water is usually admixed with solution of the gelator in a water miscible solvent. Fast gelation in presence of water which is a strong competitor for hydrogen bonds, is certainly a challenge but small molecules which can form strong gels in shorter time periods will be very valuable. To evaluate the performance of 2, its THF solution (20 mg in 0.1 mL THF) was added to 1 mL petrol in 10 mL water in a 20 mL glass vial. Remarkably the organic phase was converted to a gel in less than 30 s (ESI video S1†). When ethanol solution of 2 was used, gelation took >60 s to complete. In a larger scale (5 mL of petrol in 40 mL of water), instant gelation could be achieved with even 75 mg of the gelator in 0.5 mL THF. After separation of gel, it was taken in a RB flask and then distilled (Tg of petrol gel was 373 K) to get petrol in the receiver flask (Fig. 2). After distillation, the left-over (same Rf on TLC as of 2) was again dissolved in THF and reused two more times, with an overall efficiency of ∼70%.
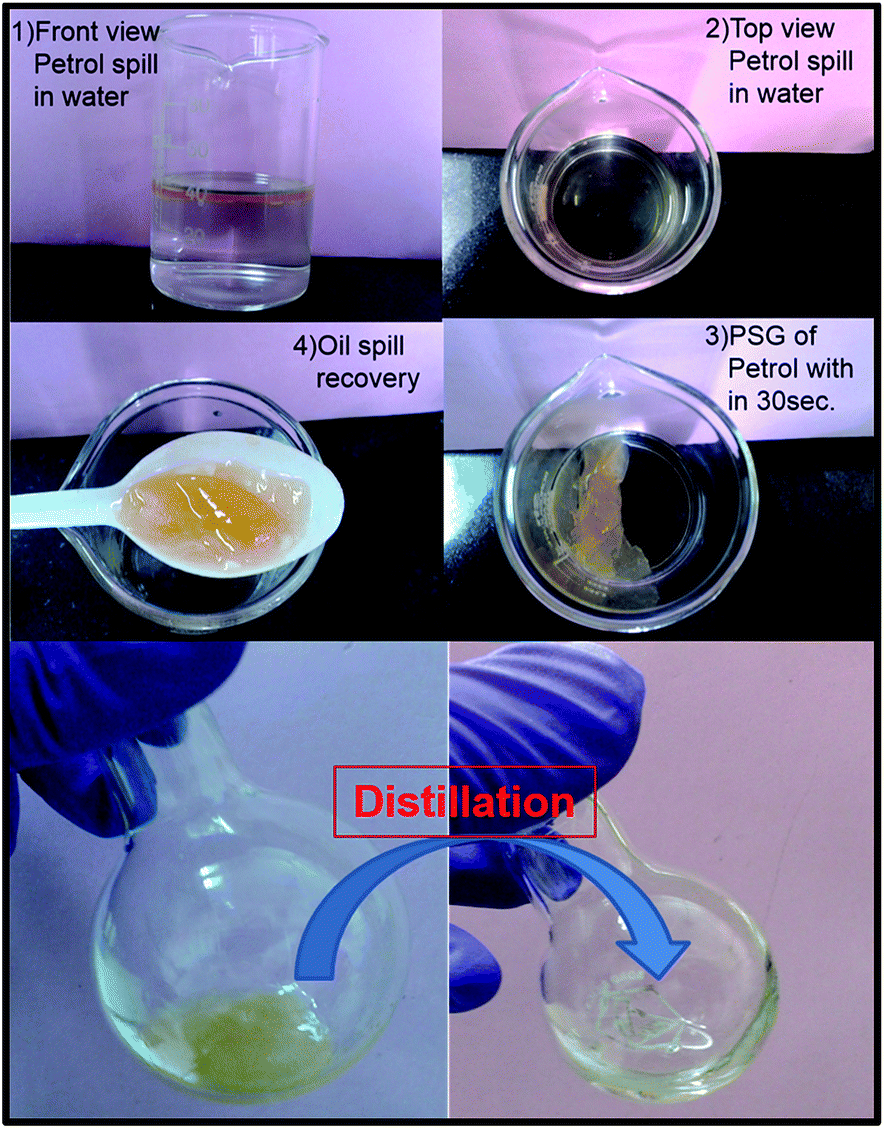 | ||
| Fig. 2 Phase selective gelation of petrol layer from a biphasic mixture of petrol and water within 30 seconds, distillation of the petrol gel to get petrol oil. | ||
Only a few reports on organogelators capable of phase-selectively gelating both oils and aromatic solvents are available.7f Since 1 and 2 belong to this category, we have explored their potential use in water decontamination, especially from organic dyes, which is a very relevant area of research. Although partitioning in aromatic solvent followed by gelation using the appropriate LMWOG is done in some cases,7f the idea of using the xerogel as a supramolecular sponge to trap the aromatic contaminants has also been demonstrated.7b,10
The gelator 2 was efficient in immobilizing dye-entrapped mesitylene in less than 30 minutes (ESI, Fig. S11 and S12†). The xerogels from aromatic solvents like mesitylene and toluene were largely fibrous as evident from the SEM images presented in Fig. 4 (also Fig. S19 and S20 ESI†). To know the difference, if any, on reversing the order, a pre-formed gel is exposed to an aqueous solution of the dye (ESI, Fig. S13 and S14†). Interestingly, irrespective of the sequence in which the gelator and the dye molecules were brought together, the nature of xerogels showed high similarity. Most notably, they were distinct from that of the xerogels formed in the absence of the dye (Fig. 4C vs. Fig. 4D). These observations suggest that there is significant interaction between the gelator and the dye molecules which cause a change in the nature of supramolecular network without adversely affecting the gelation efficiency.
 | ||
| Fig. 3 (a) Mixture of RhB and methylene blue (0.03 mM, 1 mL each), (b) mixture of both dye with mesitylene gel of 2, (c) selective trapping of RhB after 18 h, (d) UV-Vis spectrum confirming removal of RhB dye preferentially. | ||
 | ||
| Fig. 4 SEM images (A) toluene xerogel of 1, (B) toluene xerogel of 2, (C) mesitylene xerogel of 2 before dye entrapment, (D) mesitylene xerogel of 2 after dye entrapment, scale is 5 μm for (A, C and D) and 2 μm for (B). | ||
Preferential trapping of one of the dyes from their mixture in water is another characteristic that could have practical value.11 To test this, a block of mesitylene gel of 2 (1 wt%) was kept in a mixture of RhB and methylene blue (0.03 mM, 1 mL each) in water (Fig. 3a and b), and the dye entrapment was monitored by noting the change in UV absorption profile. As evident from Fig. 3c and d, both colour and absorption profile of the solution confirmed selective removal of RhB from the mixture in 18 h. Methylene blue and rhodamine B dyes are both cationic, and highly soluble in water. Preferential partitioning of the latter in aromatic solvent is attributable to its ability to equilibrate to the neutral lactone form which is not possible in the former.12
Gel strength is an important parameter that one should consider if the material is to be developed for practical application. Flow-behaviour and mechanical strength of gels are in-between solids and liquids and can be best studied by rheological experiments. The frequency- and stress sweep experiments provide the storage modulus (G′), loss modulus (G′′) and yield stress (σy) which give a good estimation of their ability to store energy as well as rigidity. Results from experiments conducted with petrol and diesel gels of 2, are clear attestation of their good rheological properties. In all cases, the G′, which is a measure of the elasticity of the material, predominated over G′′ and was nearly 10 times higher in magnitude.
In stress sweep experiment which monitors the variation of G′ and G′′ with respect to the stress amplitude (σ0) both these moduli were found to remain steady till a particular stress value and then cross each other. This point, denoted as yield stress is a measure of the rigidity of the material. Diesel and petrol gels of 2 formed at 0.5% w/v had yield stress values of 28.5 Pa, 35.7 Pa respectively (Fig. 5a). When petrol gel of 2 at 2 wt% was used, the yield stress was 269 Pa (ESI, Fig. S8†).
 | ||
| Fig. 5 (a) Oscillatory stress sweep experiment with 0.5% w/v petrol and diesel (b) angular frequency sweep experiment with 0.5% w/v petrol and diesel. | ||
As anticipated, the Tg (gel melting temperature) of mesitylene gels of 1 and 2 was found to increase with gelator concentration (ESI, Fig. S2†). Among these, strength of gel from 2 was significantly more than that from 1, indicating stable gelator–solvent interactions on having long chain at the C-terminus. Interestingly, gel from 1.5 mL of mesitylene with 0.9% w/v of 2 was able to bear the load of 250 mL of water in an inverted RB (ESI, Fig. S22†). All these show that aryl-triazolyl peptides described here are good addition to groups of soft organic materials.
During the self-assembly of peptides, hydrogen bonding from amide groups play a key role. For a better understanding of the role of such secondary interactions in the above organogelators, we performed the variable temperature 1H NMR experiment (ESI, Fig. S4 and S5†) in toluene-d8. The downfield shifting of amide protons with decrease in temperature clearly signifies the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N hydrogen bonding in both cases. A similar conclusion was also drawn from IR spectroscopic studies. The neat sample of 1 drop-casted from CHCl3 (in which 1 doesn't form gel), gave transmission bands at 3447 cm−1, 1656 cm−1 and 1510 cm−1 representing non-hydrogen bonded NH (stretching), CO (stretching) and NH (bending) vibrations. At the same time, its xerogel from toluene showed these bands at 3392 cm−1, 1630 cm−1, 1533 cm−1 respectively. The lowering of frequencies corresponding to NH & CO stretching and increase in NH bending vibrations are clear indications of H-bonded associations. A similar pattern in IR absorption bands was observed in the neat sample and xerogels of gelator 2 (ESI, Fig. S6†). In this case, the asymmetric and symmetric stretching frequencies of CH2 units in the xerogels appeared at lower wave numbers (2925 cm−1 and 2853 cm−1) in comparison with that in the neat sample prepared from CHCl3 (2927 cm−1 and 2855 cm−1) which is attributable due to better packing of long chains in the xerogels. For comparison, we have recorded IR spectrum of toluene gel of 1 and 2 (at 2 wt%) and the shift in NH and CO transmission bands were similar to those in their respective xerogels (ESI, Fig. S7†). Formation of fibrillar primary structures require 1 and 2 to adopt conformation with proper orientation of groups involved in secondary interactions. Powder XRD data of the xerogel of 1 (CCl4) and 2 (toluene) showed peaks at 2θ values of 3.66, 7.6, 10.95 and 3.5, 7.5, 10.3 respectively (d ratio fall in the order 1:1/2:1/3) indicative of layered arrangement (ESI, Fig. S17 and S18†).
O⋯H–N hydrogen bonding in both cases. A similar conclusion was also drawn from IR spectroscopic studies. The neat sample of 1 drop-casted from CHCl3 (in which 1 doesn't form gel), gave transmission bands at 3447 cm−1, 1656 cm−1 and 1510 cm−1 representing non-hydrogen bonded NH (stretching), CO (stretching) and NH (bending) vibrations. At the same time, its xerogel from toluene showed these bands at 3392 cm−1, 1630 cm−1, 1533 cm−1 respectively. The lowering of frequencies corresponding to NH & CO stretching and increase in NH bending vibrations are clear indications of H-bonded associations. A similar pattern in IR absorption bands was observed in the neat sample and xerogels of gelator 2 (ESI, Fig. S6†). In this case, the asymmetric and symmetric stretching frequencies of CH2 units in the xerogels appeared at lower wave numbers (2925 cm−1 and 2853 cm−1) in comparison with that in the neat sample prepared from CHCl3 (2927 cm−1 and 2855 cm−1) which is attributable due to better packing of long chains in the xerogels. For comparison, we have recorded IR spectrum of toluene gel of 1 and 2 (at 2 wt%) and the shift in NH and CO transmission bands were similar to those in their respective xerogels (ESI, Fig. S7†). Formation of fibrillar primary structures require 1 and 2 to adopt conformation with proper orientation of groups involved in secondary interactions. Powder XRD data of the xerogel of 1 (CCl4) and 2 (toluene) showed peaks at 2θ values of 3.66, 7.6, 10.95 and 3.5, 7.5, 10.3 respectively (d ratio fall in the order 1:1/2:1/3) indicative of layered arrangement (ESI, Fig. S17 and S18†).
In 1–3, each amino acid residue is endowed with π-stacking and hydrogen bonding groups. However, their conformations, solvation, self-assembly pattern, and hence the gelation profile, could vary depending upon the chemical environment. Kamlet and Taft have correlated the free energy of solvation with H-bond donating acidity (α), H-bond accepting basicity (β) and polarizability (π*) of solvents.13 Understandably, contribution of these parameters to the solvation energy will vary depending upon the solute–solvent combinations. In the present study, compounds 1 and 2 possessing larger number of H-bond acceptors seem to get better solvated in solvents having more H-bond donors, which is evident from their high solubility in MeOH, EtOH, CH2Cl2 and CHCl3. In isopropanol (α = 0.76, β = 0.95, π* = 0.48), these values are lower compared to methanol and ethanol (ESI, Table S2 and Fig. S3†) which favours the gelation of 1, though with a higher CGC value (2.5 wt%). Suppressing solubility by increasing the hydrophobic content of the solvent also promoted gelation as in the case of dodecanol (CGC = 2 wt%). Overall, solvents with low α, β and moderate π* values (e.g. CCl4 and mesitylene) give a balanced solubility for 1 and 2 and favour gelation. Provided α and β values are sufficiently low, even higher π* values are tolerated (e.g. bromobenzene).
In the present report, we have introduced a new, easily accessible synthetic peptide with possibility of fine-tuning the gelation characteristics through C-terminal modification. Optimal flexibility conferred by the aryltriazolyl- and amide units in each residue and their participation in the stabilization of extended assembly allowed us to get the gels of a wide variety of solvents (aromatic & aliphatic). Ability to: (i) phase selectively gelate petrol in presence of water, (ii) decontaminate water from a dye such as rhodamine B, and (iii) good rheological properties (σy = 269 Pa for petrol gel), are the other notable observations.
Acknowledgements
Financial support of this work by the Department of Science and Technology, INDIA (SR/NM/NS-1051/2012(G)), instrumentation facility by IIT Madras, and DST-FIST (New Delhi) are gratefully acknowledged. We thank Professor Abhijeet Deshpande, Department of Chemical Engineering, IIT Madras for Rheological studies. We also thank Department of Metallurgical and Materials Engineering, IIT Madras, SAIF IIT Madras for SEM facilities. BKS thanks CSIR, New Delhi, India for fellowship.Notes and references
- (a) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC; (b) R. G. Weiss and P. Terech, Materials with Self-Assembled Fibrillar Networks, Springer, Dordrecht, 2006 Search PubMed; (c) J. W. Steed, Chem. Commun., 2011, 47, 1379–1383 RSC; (d) B. Escuder and F. Juan, Miravet Functional Molecular Gels, The Royal Society of Chemistry, Cambridge, 2014 Search PubMed; (e) D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237 CrossRef CAS; (f) A. Vintiloiu and J. C. Leroux, J. Controlled Release, 2008, 125, 179–192 CrossRef CAS PubMed; (g) A. Vidyasagar, K. Handore and K. M. Sureshan, Angew. Chem., Int. Ed., 2011, 50, 8021–8024 CrossRef CAS PubMed; (h) S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698 CrossRef CAS PubMed.
- (a) C. Tomasini and N. Castellucci, Chem. Soc. Rev., 2013, 42, 156–172 RSC; (b) R. Afrasiabi and H. B. Kraatz, Chem.–Eur. J., 2013, 19, 15862–15871 CrossRef CAS PubMed; (c) T. Kar, S. Mukherjee and P. K. Das, New J. Chem., 2014, 38, 1158–1167 RSC; (d) A. Banerjee, G. Palui and A. Banerjee, Soft Matter, 2008, 4, 1430–1437 RSC; (e) T. Kar, S. Debnath, D. Das, A. Shome and P. Das, Langmuir, 2009, 25, 8639–8648 CrossRef CAS PubMed; (f) C. W. Liu, M. Su, X. L. Li, T. Xue, N. Liu, J. Yin, Y. Y. Zhu and Z. Q. Wu, Soft Matter, 2015, 11, 5727–5737 RSC; (g) C. G. Pappas, P. W. J. M. Frederix, T. Mutasa, S. Fleming, Y. M. Abul-Haija, S. M. Kelly, A. Gachagan, D. Kalafatovic, J. Trevino, R. V. Ulijn and S. Bai, Chem. Commun., 2015, 51, 8465–8468 RSC; (h) M. Konda, I. Maity, D. B. Rasale and A. K. Das, ChemPlusChem, 2014, 79, 1482–1488 CrossRef CAS; (i) Z. Ke, H. F. Chow, M. C. Chan, Z. Liu and K. H. Sze, Org. Lett., 2012, 14, 394–397 CrossRef CAS PubMed.
- (a) S. Datta and S. Bhattacharya, Chem. Soc. Rev., 2015, 44, 5596–5637 RSC; (b) J. Tritt-Goc and J. Kowalczuk, Langmuir, 2012, 28, 14039–14044 CrossRef CAS PubMed; (c) J. Cui, A. Liu, Y. Guan, J. Zheng, Z. Shen and X. Wan, Langmuir, 2010, 26, 3615–3622 CrossRef CAS PubMed; (d) G. John, G. Zhu, J. Li and J. S. Dordick, Angew. Chem., Int. Ed., 2006, 45, 4772–4775 CrossRef CAS PubMed; (e) X. Guan, K. Fan, T. Gao, A. Ma, B. Zhang and J. Song, Chem. Commun., 2016, 52, 962–965 RSC.
- (a) M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS PubMed; (b) H. Yu, B. Liu, Y. Wang, J. Wang and Q. Hao, Soft Matter, 2011, 7, 5113–5115 RSC; (c) N. Vidhya Lakshmi, T. Madhu Babu and E. Prasad, Chem. Commun., 2016, 52, 617–620 RSC.
- J. P. Desvergne, Beilstein J. Org. Chem., 2010, 6, 846–847 CrossRef CAS PubMed.
- (a) M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC; (b) C. Wang, Q. Chen, F. Sun, D. Zhang, G. Zhang, Y. Huang, R. Zhao and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3092–3096 CrossRef CAS PubMed; (c) Y. Jiang, F. Zeng, R. Gong, Z. Guo, C. F. Chen and X. Wan, Soft Matter, 2013, 9, 7538–7544 RSC; (d) R. Rajaganesh, A. Gopal, T. Mohan Das and A. Ajayaghosh, Org. Lett., 2012, 14, 748–751 CrossRef CAS PubMed; (e) J. A. Foster, M. PiepenbrockMarc-Oliver, G. O. Lloyd, N. Clarke, A. K. HowardJudith and J. W. Steed, Nat. Chem., 2010, 2, 1037–1043 CrossRef CAS PubMed; (f) Q. Lin, T. T. Lu, X. Zhu, B. Sun, Q. P. Yang, T. B. Wei and Y. M. Zhang, Chem. Commun., 2015, 51, 1635–1638 RSC; (g) W. Miao, L. Qin, D. Yang, X. Jin and M. Liu, Chem.–Eur. J., 2015, 21, 1064–1072 CrossRef CAS PubMed; (h) P. Rajamalli and E. Prasad, Org. Lett., 2011, 13, 3714–3717 CrossRef CAS PubMed.
- (a) S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185–186 RSC; (b) S. Mukherjee and B. Mukhopadhyay, RSC Adv., 2012, 2, 2270–2273 RSC; (c) S. Mukherjee, C. Shang, X. Chen, X. Chang, K. Liu, C. Yu and Y. Fang, Chem. Commun., 2014, 50, 13940–13943 RSC; (d) S. Basak, J. Nanda and A. Banerjee, J. Mater. Chem., 2012, 22, 11658–11664 RSC; (e) C. C. Tsai, Y. T. Cheng, L. C. Shen, K. C. Chang, I. T. Ho, J. H. Chu and W. S. Chung, Org. Lett., 2013, 15, 5830–5833 CrossRef CAS PubMed; (f) Y. Zhang, Y. Ma, M. Deng, H. Shang, C. Liang and S. Jiang, Soft Matter, 2015, 11, 5095–5100 RSC; (g) L. Yan, G. Li, Z. Ye, F. Tian and S. Zhang, Chem. Commun., 2014, 50, 14839–14842 RSC; (h) A. Prathap and K. M. Sureshan, Chem. Commun., 2012, 48, 5250–5252 RSC.
- (a) D. Balamurugan and K. M. Muraleedharan, Chem.–Eur. J., 2012, 18, 9516–9520 CrossRef CAS PubMed; (b) D. Balamurugan and K. M. Muraleedharan, Soft Matter, 2012, 8, 11857–11862 RSC; (c) D. Balamurugan and K. M. Muraleedharan, Chem.–Eur. J., 2015, 21, 9332–9338 CrossRef CAS PubMed.
- (a) R. M. Atlas and T. C. Hazen, Environ. Sci. Technol., 2011, 45, 6709–6715 CrossRef CAS PubMed; (b) R. R. Lessard and G. Demarco, Spill Sci. Technol. Bull., 2000, 6, 59–68 CrossRef CAS.
- S. Debnath, A. Shome, S. Dutta and P. K. Das, Chem.–Eur. J., 2008, 14, 6870–6881 CrossRef CAS PubMed.
- N. Cheng, Q. Hu, Y. Guo, Y. Wang and L. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 10258–10265 CAS.
- R. W. Ramette and E. B. Sandell, J. Am. Chem. Soc., 1956, 78, 4872–4878 CrossRef CAS.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, mass analysis, gelation studies etc. See DOI: 10.1039/c6ra01282e |
| This journal is © The Royal Society of Chemistry 2016 |