
First principles study on the structural evolution and properties of (MCl)n (n = 1–12, M = Cu, Ag) clusters
Zhimei Tianab and
Longjiu Cheng*a
aDepartment of Chemistry, Anhui University, Hefei, Anhui 230601, China. E-mail: clj@ustc.edu
bSchool of Chemistry and Materials Engineering, Fuyang Teachers College, Fuyang, Anhui 236037, China
First published on 14th March 2016
Abstract
The structural evolution of (MCl)n (M = Cu, Ag, n = 1–12) clusters has been explored using a first principles global minimization technique, namely, a genetic algorithm from density functional theory geometry optimization (GA-DFT). Benchmark calculations indicate that the method used in this work is reliable in predicting the energetic sequences of different isomers of (CuCl)6 clusters compared to the high-level coupled cluster method. The computational results indicate that the structures of (CuCl)n and (AgCl)n clusters share many similarities. The global minima of (MCl)n clusters are planar and nonplanar monocyclic structures up to n = 5, and multi-ring or catenane structures when n ≥ 6. The characters of the intramolecular interactions of (AgCl)3, (CuCl)3, (CuCl)4 and (CuCl)6 clusters have been studied by the noncovalent interactions (NCI) index. The electronic and bonding properties of (CuCl)3 and (AgCl)3 clusters are analyzed. At a higher level, the refined energies of the isomers, the binding energies and the second difference of binding energies are calculated, and are examined as a function of the cluster size. Several promising sizes of higher stability and symmetry have been discovered. In particular, we find that the cluster adopts a new folding manner, tube conformation, at n = 6, 9 and 12, where they are more stable than the adjacent clusters; moreover, they are aromatic.
Introduction
The study of coinage metal halide clusters has attracted much attention in experiments1,2 and computations3–5 because of their practical uses, such as in catalysis and photography, etc. From a more fundamental point of view, the interactions between halide and noble metal atoms are difficult to characterize. The ionic bonding character is less marked than in alkali halide systems and the presence of d electrons of the noble metal makes theoretical investigations much more complicated.6 The exploration of cluster structure is the first step to understand the properties of bulk material. The group 11 halides are particularly interesting. Relativistic effects are important for group 11 compounds, especially for gold compounds, which can lead to significant changes in chemical properties.7–10 Closed-shell d10⋯d10 interactions among the coinage metal halide clusters have also attracted considerable interest.11,12The crystal structure of CuCl and AgCl have been synthesized,13–15 however, the physical and chemical properties of small clusters often exhibit significant differences with bulk phase. The evolution of structures and properties with cluster size is a major challenge for experimental and theoretical study.16 The structures, bonding of Ag2X, AgX, AgX2, and AgX3 (X = F, Cl, Br, I) in gas-phase have been studied.17 The structure and bonding in MnXn (M = Cu, Ag, Au; X = Br, I; n = 1–6), have been reported.18 The trimer evaporation in silver bromide clusters have been used to study the stability, structural and electronic properties of (AgnBrp)(+) (n = 6, p = n, n − 1) clusters.19 The trimer formation of AgI has been studied experimentally and theoretically.20 The structures and properties of (AgBr)n (n ≤ 6) have been studied.16 The molecular and electronic structures of (AgBr)n (n = 1–9) clusters have been investigated.21 The band structures of AgF, AgCl, and AgBr have been reported.22 A comparison of structure and stability between M4X4 (M = Cu, Ag, Au; X = F, Cl, Br, I) has been performed23 and similar cyclic structures are obtained. Structure and stability of (CuCl)n and (AgCl)n (n = 1–6) clusters have been studied.6 Walsh et al. predicted the existence and chemical stability of cuprous fluoride.24 The ground state structures of (CuCl)n and (AgCl)n (n = 4–6) in previous calculations were not in consistent with each another.6,23 The possible nuclear arrangements of (CuCl)n and (AgCl)n were guessed in previous computations. Thus, the results from the guessed structures are biased. It is worth mentioning that our initial structures of (CuCl)n and (AgCl)n are not guessed, but from the results of genetic algorithm (GA).25–27 The initial structures of GA are then optimized using DFT method.
As former reports reveal, the (MX)3 cluster is particular stable, however, there only exist interpreting analysis of the molecular orbitals and ionization energy of the cluster.4,6,16 Compounds containing two or more metal atoms separated by a shorter distance than the sum of their covalent radii, termed a metallophilic contact such as aurophilic, cuprophilic and argentophilic contacts, are of great interest in both academia and industry.28–31 These interactions are found to influence significantly a variety of structural and physical characteristics of gold, silver and copper compounds. Lots of experimental and computational studies have been dedicated to the metallophilic contact phenomenon. The noncovalent interaction (NCI) index32 is applied to characterize the phenomenon of metallophilic interactions.11,28,33,34 Clearly, a much better understanding of the electronic structure and interactions within the cuprophilic and argentophilic contacts region is necessary for the (MX)3 cluster.
To the best of our knowledge, there are few reports on (MCl)n (M = Cu, Ag) clusters with n > 6. Here we report a systematic study of the structures and properties of (MCl)n (M = Cu, Ag, n = 1–12) clusters. The structures of the clusters are obtained; the stability, noncovalent interactions and aromatic properties are calculated and discussed.
Computational details
Computational methods
The structures of (MCl)n (M = Cu, Ag, n = 1–12) clusters are calculated with the combination of GA and DFT method, which has been successfully applied in the structural predictions of many systems.26,35–39 GA is a search heuristic that mimics the process of natural selection.25,40 This heuristic is routinely used to generate useful solutions to optimization and search problems. It uses operators that are analogues of the evolutionary processes of mating (or “cross-over” at the gene level), mutation and natural selection to explore multi-dimensional parameter spaces.26 The GA belongs to the class of evolutionary algorithms, which is an optimization strategy inspired by the Darwinian evolution. This strategy is routinely used to generate useful solutions to optimization and search problems. Starting with a population of candidate structures, we relax these candidates to the nearest local minimum. Using the relaxed energies as the criteria, a fraction of the population is selected as “parents”. The next generation of candidate structures is the structure mating the “parents”. The progress is repeated until the global minimum (GM) is located.25,41 DFT based GA is very time-consuming, and when cluster size n ≥ 2, more than 1000 samplings are optimized by DFT. The quantum chemical calculations are performed with the Gaussian 09 suite of programs.42 In the optimization procedure, the Lanl2dz basis set and 6-31G(d) basis set are used for the coinage metals and Cl at TPSSh functional level43 without any symmetry constraint, and the normal mode frequencies are also computed at the same level for all structures to ensure that they belong to minima. All the minima are verified by the absence of imaginary frequency. The refined energies of the isomers and the binding energies are calculated using the small-core relativistic effective core potentials, ECP10MDF43 and ECP28MDF44 for Cu and Ag, respectively, with the corresponding correlation consistent valence basis sets (aVDZ),44,45 and the aug-cc-pvdz basis set for Cl. Hereafter, the reported energy differences of the isomers are calculated at this level. The ionization potentials of Cu and Ag atoms calculated at this level are 7.80 and 7.67 eV, respectively, which are in consistent with the corresponding experimental values (7.73 and 7.58 eV),46 indicating that our calculation method is reliable. Noncovalent interaction (NCI) plots are plotted using Multiwfn47,48 and VMD49 packages. The nucleus-independent chemical shift (NICS)50 method has been used to study the aromaticity of partial clusters.Benchmark calculations
To verify the reliability of the methods used in this work, a benchmark calculation is carried out by comparing the relative stability of the three isomers of (CuCl)6 cluster (see structures in Fig. 1) using different basis sets at CCSD(T) level of theory. Results of the benchmark calculations are given in Table 1. Note that the relative stabilities of 6II and 6III are highly overestimated by TPSSh/Lanl2dz and 6-31G(d) method. However, the relative stability of the three isomers obtained by the refined energies at TPSSh level is in overall consistent with the results obtained at the CCSD(T) level. The comparison suggests that the refined energies are necessary and reliable in predicting relative stability of (CuCl)6 cluster.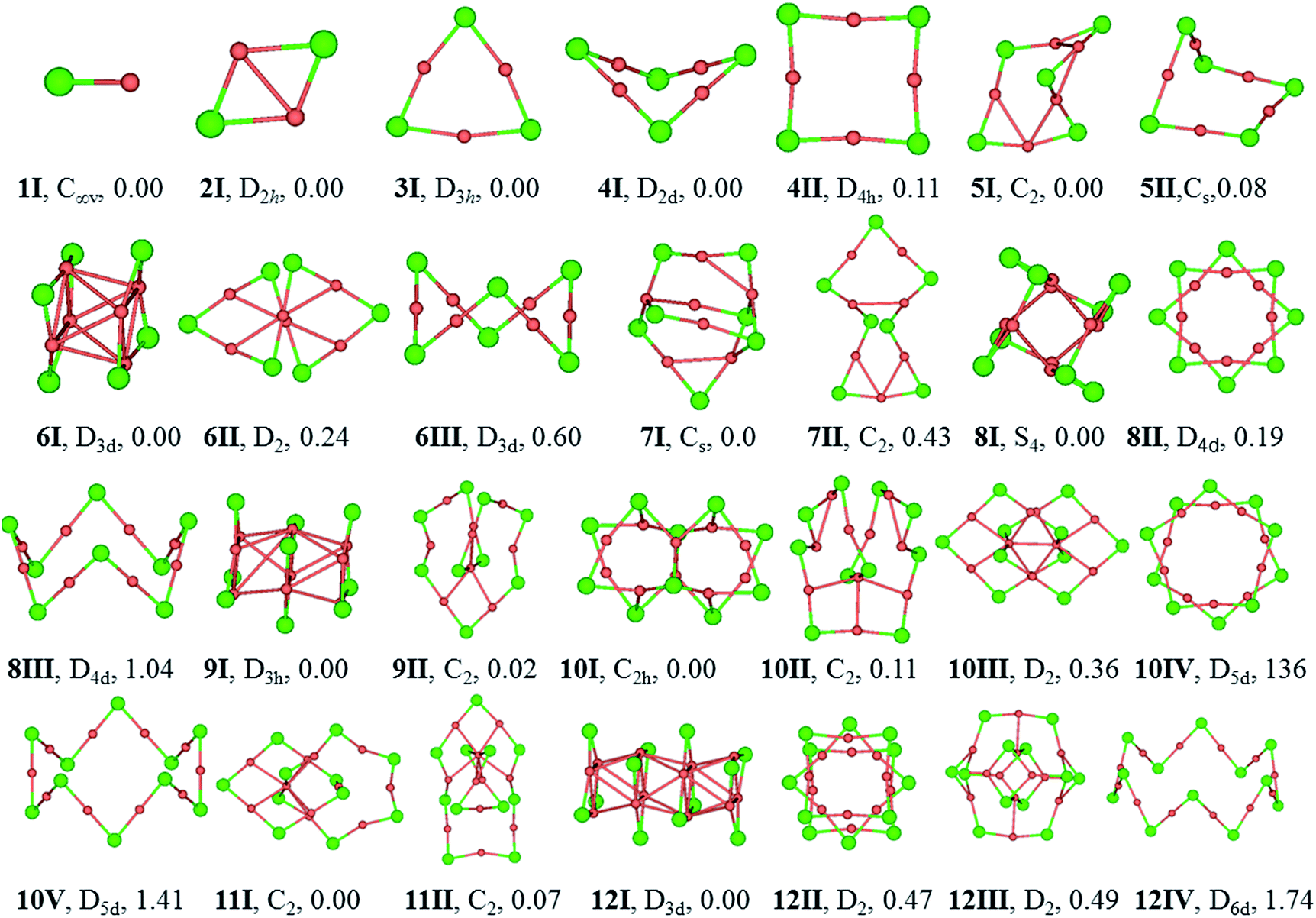 | ||
| Fig. 1 Optimized structures of low-lying isomers of (CuCl)n (n = 1–12) at TPSSh/Lanl2dz (Cu) and 6-31G(d) (Cl) level of theory. The numbers under the structures are the relative energies in eV at TPSSh level using aVDZ and aug-cc-pvdz basis sets for Cu (ECP10MDF) and Cl, respectively. The structures labelled in nI correspond to the global minima, nII–nV are local minima that are higher in energy, and the symmetries are labeled in figure. Cu-pink, Cl-green. | ||
| Method | 6I | 6II | 6III |
|---|---|---|---|
| a Energies for 6I are in atomic units, while other energies are relative to this in eV. Results are single point energies for TPSSh (Lanl2dz (metal) and 6-31G(d) (Cl)) geometry.b Energies are calculated using the relativistic effective core potential ECP10MDF for Cu with the corresponding correlation consistent valence basis sets (aVDZ), and the aug-cc-pvdz basis set for Cl. | |||
| TPSSh/Lanl2dz, 6-31G(d) | −3938.0677611 | 0.75 | 1.15 |
| TPSShb | −3945.4014317 | 0.13 | 0.46 |
| CCSD(T)/aug-cc-pvdz, aug-cc-pvdz-pp | −3938.4000069 | 0.02 | 0.20 |
| CCSD(T)/Lanl2dz | −1260.0751833 | 0.20 | 0.15 |
| CCSD(T)/6-311+G(d) | −12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 594.0672619 594.0672619 |
0.23 | 0.34 |
| CCSD(T)/def2-tzvp | −12594.8747943 |
0.11 | 0.14 |
| CCSD(T)/cc-pvtz | −12595.7453668 |
0.03 | 0.05 |
Results and discussion
The equilibrium geometries of (CuCl)n and (AgCl)n with n = 1–12 clusters are plotted in Fig. 1 and 2, respectively. The structures are indexed in Roman number order (I, II, III) and alphabetical order (A, B, C) by the energy from low to high for (CuCl)n and (AgCl)n clusters, respectively. The symmetry and relative energies of the isomers are also shown.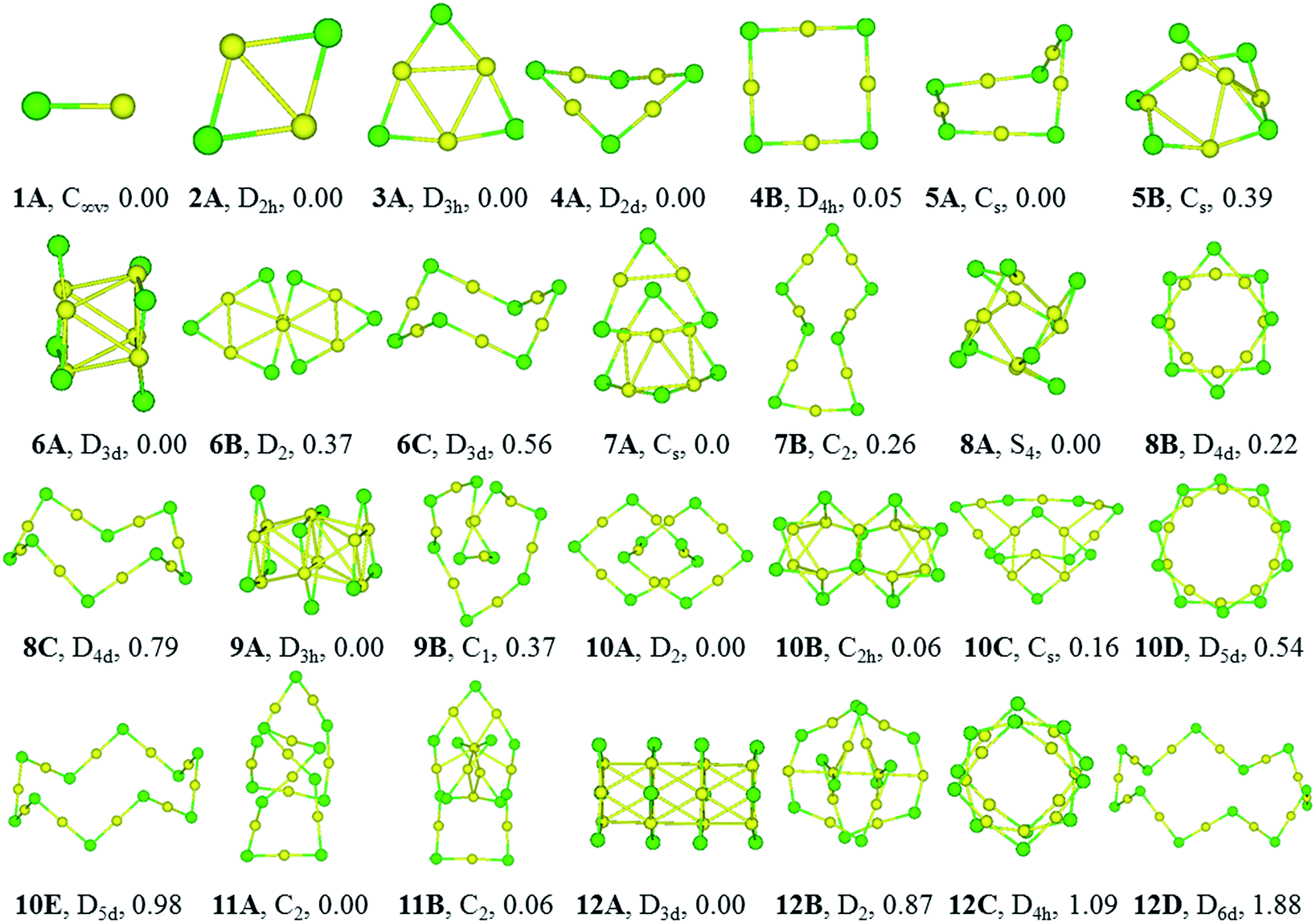 | ||
| Fig. 2 Optimized structures of low-lying isomers of (AgCl)n (n = 1–12) at TPSSh/Lanl2dz (Ag) and 6-31G(d) (Cl) level of theory. The numbers under the structures are the relative energies in eV at TPSSh level using aVDZ and aug-cc-pvdz basis sets for Ag (ECP28MDF) and Cl, respectively. The structures labelled in nA correspond to the global minima, nB–nE are local minima that are higher in energy, and the symmetries are labeled in figure. Ag-yellow, Cl-green. | ||
Geometric structure
The bond length of 1I and 1A are 2.11 and 2.34 Å, respectively, which are both in C∞v symmetry. The corresponding experimental results are 2.055 and 2.281 Å.51,52 Both of them are reasonably close to the experimental values.For dimers, the D2h rhombus is the lowest-energy isomer, namely, 2I and 2A. In 2I, the Cu–Cl and Cu–Cu bond lengths are 2.32 and 2.44 Å, respectively, and a Cu–Cl–Cu angle of 63°. The Ag–Cl and Ag–Ag distances in 2A are 2.56 and 2.85 Å, respectively, and Ag–Cl–Ag angle of 68°. Our results are in accordance with similar system M2Br2, M = Cu, Ag and Au using B3LYP functional.18
The trimer (3I and 3A) is a quasi-triangular shape with D3h symmetry and strong M–M interactions favored by relatively short distances (2.65 and 2.99 Å for Cu–Cu and Ag–Ag bond lengths, respectively). 3I and 3A consist of halide atoms with metal atoms bridging the Cl–Cl bonds. The Cu–Cl bond lengths in 3I are all equivalent, 2.24 Å. The Ag–Cl bond lengths in 3A are also the same, 2.46 Å. In agreement with previous B3LYP and matrix-IR spectra reports of (AgCl)3, our calculations give a planar six-membered ring structure.20,53 The Cl–Cu–Cl and Cl–Ag–Cl angles in 3I and 3A are 167° and 165°, respectively. The Cu–Cl–Cu and Ag–Cl–Ag angles are 73° and 75° in 3I and 3A, respectively.
For tetramer, it favors a nonplanar distorted D2d symmetry structure (4I and 4A) with a D4h-symmetry isomer (4II and 4B) 0.11 and 0.05 eV higher in energy for (CuCl)4 and (AgCl)4, respectively. The Cu–Cl and Cu–Cu bond distances in 4I are 2.21 and 2.76 Å, respectively, and the Cl–Cu–Cl angle is 178°, which indicates that the ClCuCl atoms are almost aligned. The Ag–Cl and Ag–Ag bond distances in 4A are 2.43 and 3.24 Å, respectively, and the Cl–Ag–Cl angle is 179°. In 4B, the Ag–Cl and Ag–Ag distances are 2.43 and 3.46 Å, respectively, the Cl–Ag–Cl and Ag–Cl–Ag angles are 179° and 91°, respectively. Our computational results of the stable isomers are in agreement with that of (CuBr)4 cluster.54
5I and 5A are in cyclic arrangement, which are in C2 and Cs symmetries, respectively. It can be described as a nonplanar distorted pentagon with Cl apexes and ClMCl sides having different M–Cl–M angles (ranging from 71° to 98°) around the cycle. The 10 M–Cl bond lengths are in the range of 2.22–2.27 and 2.43–2.44 Å for Cu–Cl and Ag–Cl, respectively. 5II is a distorted structure with Cs symmetry, 0.08 eV higher in energy than 5I. 5B is a Cs symmetry structure with one chloride atom capping one tetramer ring, 0.39 eV higher in energy than 5A.
6I and 6A are both tube structures in D3d symmetry with two quasi-planar triangle units paralleling to each other. The conformation has been revealed in (AgBr)6 cluster.16 The 12 M–Cl bond lengths are strictly identical in 6I and 6A, 2.29 and 2.52 Å for Cu–Cl and Ag–Cl, respectively, and the metal–metal distances are found to be 2.72 and 3.08 Å for Cu–Cu and Ag–Ag in the two triangles of 6I and 6A, respectively. 6II and 6B are helix structures in D2 symmetry, lying 0.24 and 0.37 eV higher in energy than 6I and 6A, respectively. 6III and 6C are the known zigzag crown conformations in D3d symmetry, lying 0.60 and 0.56 eV higher in energy than 6I and 6A, respectively.
7I and 7A are distorted structures in Cs symmetry, consisting of one six-member-ring and one eight-member-ring. The Cu–Cl bond lengths are in the range of 2.21–2.58 Å and Cl–Cu–Cl angles are in the range of 103.4–164.1° in 7I. In 7A, the Cl–Ag–Cl angle that near the center of the (AgCl)4 part is 157.4°, and much smaller than the two adjacent Cl–Ag–Cl angles (both 166.6°) in the triangle, which perhaps because it helps to stabilize the structure. 7II and 7B are monocyclic structure in C2 symmetry, 0.43 and 0.26 eV higher in energy than 7I and 7A, respectively.
8I and 8A are windmill structures in S4 symmetry, and both of which are composed of four seven-member-ring and two six-member-ring units. The Cl–M–Cl angles are in the range of 118.4–163.9°. In 8I, the bond lengths of Cu–Cl are in the range of 2.24–2.42 Å and the Cu⋯Cu distance is 2.67 Å; in 8A, the bond lengths of Ag–Cl are in the range of 2.47–2.64 Å and the Ag⋯Ag distance is 3.03 Å. 8II and 8B in D4d symmetry are separate-ring structures including two paralleling tetramer units, 0.19 and 0.22 eV higher than 8I and 8A, respectively. 8III and 8C are zigzag crown conformations in D4d symmetries, lying 1.04 and 0.79 eV higher in energy than 8I and 8A, respectively.
9I and 9A are both tube structures in D3h symmetry with three triangular units paralleling to each other. It can be viewed as a tube from the center of the triangular units. In 9I and 9A, the 12 M–Cl bond lengths are strictly identical in the side triangles, 2.29 and 2.51 Å, and the 6 M–Cl bond lengths are 2.39 and 2.59 Å in the middle triangle; the M–M bond between the triangle units are strictly 2.84 and 3.19 Å, respectively. 9II and 9B are helical structures, lying 0.02 and 0.37 eV higher in energy than 9I and 9A, respectively.
10I and 10B are perfect C2h symmetry structures, consisting of intersecting triangular units. 10A is a catenane structure in D2 symmetry. Worth noting is that (AuSR)10 cluster has been synthesized and characterized as catenane structure.55 In 10I, the Cu–Cl bond lengths range from 2.24 to 2.42 Å, and the Cu–Cl–Cu angle in the center of the structure is 81.2°, which deviates the linearity seriously. 10II is a helix structure, 0.11 eV higher in energy than 10I. 10III is a catenane structure, lying 0.36 eV higher than 10I. 10C is a Cs structure consisting of two rings inserting into each other. 10IV and 10D are separate-ring structures consisting of two pentamer units. 10V and 10E are crown structures in D5d symmetries, 1.41 and 0.98 eV higher in energy than 10I and 10A, respectively.
11I and 11A are catenane structures in C2 symmetry, with interlocked hexamer and pentamer rings. The Cu–Cl bond lengths in 11I are in the range of 2.21–2.34 Å, and Cl–Cu–Cl angles are in the range of 148.9–174.3°. The Ag–Cl bond lengths in 11A are in the range of 2.43–2.56 Å, and ClAgCl angles range from 160.2 to 178.5°. 11II and 11B compete with 11I and 11A only 0.07 and 0.06 eV higher in energy, respectively, with a tetramer inserting into a heptamer.
12I and 12A are both tube structures in D3d symmetry, including four paralleling triangle units. The Cu–Cl bond lengths in the side triangles are 2.29 Å and those in the middle triangles are 2.39 Å, and Cu–Cu bonds are in the range of 2.81–2.87 Å in 12I. The Ag–Cl bond lengths are strictly identical in the side triangles (2.51 Å), whereas those in the two middle triangles are all 2.59 Å in 12A. 12II and 12B are 0.47 and 0.87 eV higher in energy than 12I and 12A, respectively. 12II and 12C are both separate ring structures in D4h symmetry, including three tetramer ring units. 12III is a three-dimensional structure and 12B is a catenane structure. 12IV and 12D are zigzag crown D6d structures, lying 1.74 and 1.88 eV higher in energy than 12I and 12A, respectively.
The stable structures in the literatures are all included in our results of GA. The (MCl)n clusters are planar or nonplanar cycles up to n = 5, which is in consistent with similar system (AgBr)n.19 In this work, the D3d (AgCl)6 isomer is the GM, whereas that of a zigzag crown C3v symmetry structure cluster is the GM of (AgBr)6. The zigzag structure in our work converges to the D3d isomer.19 Moreover, our results are in agreement with more recent work about (MBr)4 (M = Cu, Ag and Au).18 The most stable isomers of (AgCl)5 and (AgCl)6 are in Cs and D3d symmetries, which are different from those of (AgF)5 and (AgF)6 whose lowest-energy isomers are in D5h and D6h symmetries.2 The reason for this is probably due to the strong ionic bond characterization between fluorine and silver atoms. The (AgF)n are planar cycles up to n = 6, whereas the (MCl)n (M = Cu and Ag) systems can keep planar or non-planar cycles only up to n = 5, which suggests that the halogen might be responsible for the tendency to become non-planar.4 The structural patterns of (CuCl)n and (AgCl)n clusters are in consistent with that of (AuCl)n clusters when n = 1–5, however, they are significantly different from (AuCl)n clusters when n > 5 due to the strong aurophilicity of Au element.56
Stability
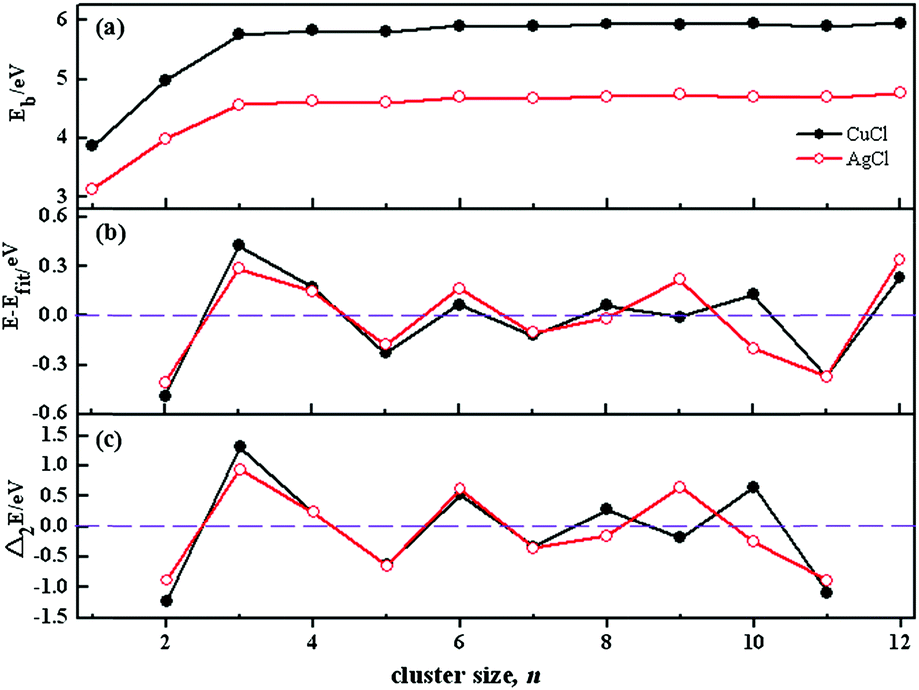 | ||
| Fig. 3 (a) Binding energies for (CuCl)n and (AgCl)n clusters as a function of cluster size n; (b) plot of the energetic gaps (E − Efit) of the GMs for (CuCl)n and (AgCl)n clusters as a function of cluster size n, where E is the total energy, and Efit is a four-parameter fit of the GMs. (CuCl)n clusters: Efit = 3.57471 + 1.17587 × n1/3 − 0.40799 × n2/3 + 0.05064 × n, (AgCl)n clusters: Efit = 3.57466 + 1.17591 × n1/3 − 0.40800 × n2/3 + 0.05063 × n; (c) second difference of binding energies for (CuCl)n and (AgCl)n clusters as a function of cluster size n. | ||
The stability of these clusters is also established by the energetic gaps (E − Efit) of the GMs, where E is the total energy and Efit is a four-parameter fit of the GM, respectively. The relative stability of the clusters is further assessed with the second difference of binding energies from the following formula: Δ2Eb(n) = Eb(n + 1) + Eb(n − 1) − 2Eb(n), where Eb is the total binding energy divided by n. The second difference of binding energies is previously used to predict the relative stability of other clusters.57,58 E − Efit and Δ2E(n) are calculated in a manner that emphasizes particular stable minima or “magic numbers” of the clusters. In such curves, positive peaks correspond to more stable structures. The plots of E − Efit and Δ2E(n) as a function of cluster size are shown in Fig. 3b and c, respectively. It can be clearly seen from the figures that, distinctive peaks present at cluster sizes 3, 6, 9 and 12 for (AgCl)n clusters, indicating their higher relative stability than their neighbours. However, there are distinctive peaks at cluster sizes 3, 6, 8, 10 and 12 of (CuCl)n clusters. The reasons for the stability difference between (CuCl)n and (AgCl)n clusters may be that the GMs at n = 10 are different from each other. The particular stability of (AgCl)3 is in agreement with that of (AgBr)3, which is previously reported experimentally and theoretically.1,3,21
and the representation of s versus ρ shows characteristic peaks at low density values in the presence of noncovalent interactions. The sign of the second eigenvalue (λ2) of the electron-density Hessian matrix is used to distinguish between bonded (λ2 < 0) and non-bonded (λ2 > 0) interactions. Since the sign of the second eigenvalue sign (λ2) is indicative for the type of interaction, the analysis results are shown in scatter plots of the reduced density gradient (s) versus the electron density (ρ) multiplied by the sign of λ2. The spikes in low-density, low-gradient regions in the plots of s versus sign (λ2)ρ exhibit the noncovalent interactions. The visualization of the gradient isosurface can be seen in real space through VMD program.49 The gradient isosurfaces are colored according to the corresponding values of sign (λ2)ρ, which is found to be a good indicator of interaction strength.32 A RGB (red-blue-green) scale is used.
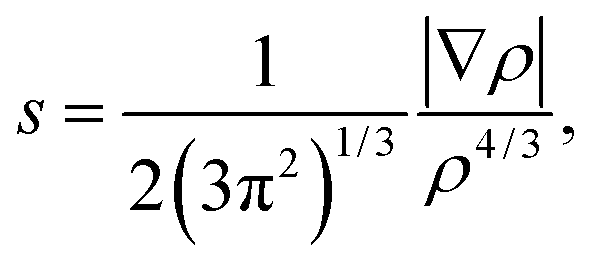
The NCI analysis for (AgCl)3, (CuCl)3, (CuCl)4 and (CuCl)6 clusters is depicted in Fig. 4. Several regions of intramonomer interactions can be distinguished. Favourable (attractive bonding) interactions appear on the left, while unfavourable (nonbonding) on the right. The strength of the NCI is proportional to the density on the NCI surface. Fig. 4a shows the bicolored isosurfaces of (AgCl)3 and (CuCl)3 clusters. The weakening of the metal–metal interaction on going from Cu to Ag (Fig. 4a) can be simply monitored thanks to the shift of the characteristic peak towards more negative values in the NCI plot. The results in Fig. 4a reveal that one strong attractive spike (−0.023 a.u.) is clearly associated to the Ag⋯Ag interactions in (AgCl)3, whereas two strong steric repulsive spikes (0.016 and 0.023 a.u. of sign (λ2)ρ) located at the center and near the M–M bonds of (AgCl)3 cluster. Similarly, there are one attractive spike (−0.029 a.u.) associated to Cu⋯Cu interactions, and two repulsive spikes (0.025 and 0.029 a.u. of sign (λ2)ρ) for (CuCl)3. The attractive and repulsive interactions are clearly stronger than those in (AgCl)3 according the values of sign (λ2)ρ. The isosurfaces of (CuCl)3 and (AgCl)3 in the right part of Fig. 4a reveal that the attractive interactions are in blue, whereas the repulsive interactions are in red. It is worth mentioning that the Cu⋯Cu interactions in (CuCl)3 is stronger than the H bond in water dimer (−0.025 a.u.).32
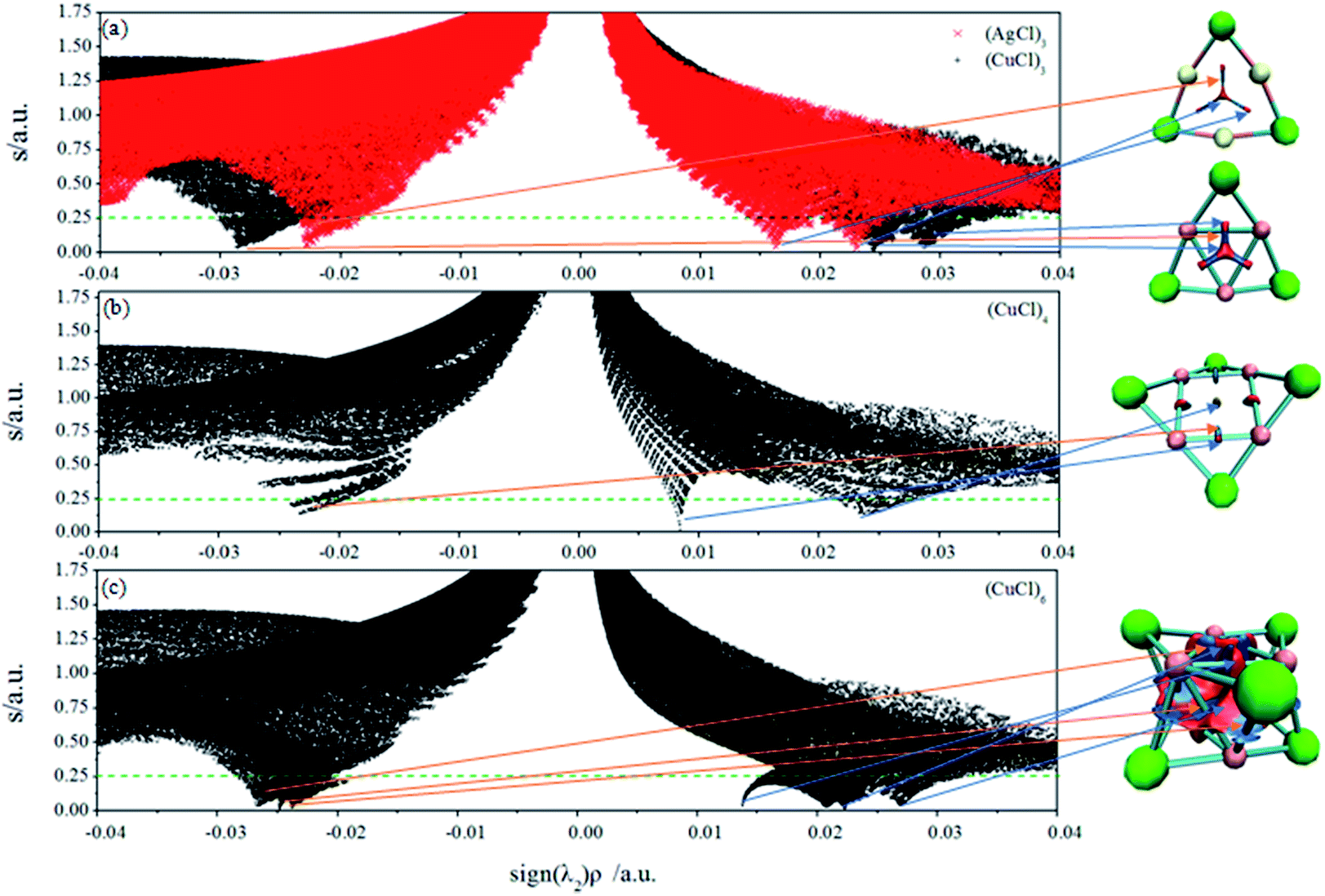 | ||
| Fig. 4 Plots of the reduced density gradient versus the electron density ρ multiplied by the sign of the second Hessian eigenvalue sign (λ2) (left) and NCI isosurfaces using a blue-green-red colour scale at s = 0.25 for the global minim structures (right) of (a) (AgCl)3 and (CuCl)3, (b) (CuCl)4 and (c) (CuCl)6 clusters, respectively. Results were obtained on a cuboid grid using the density and gradient values at the TPSSh/Lanl2dz/6-31G(d) level of theory. Ag-yellow, Cu-pink, Cl-green. | ||
The NCI analysis and isosurface (Fig. 4b) of (CuCl)4 cluster are used to demonstrate the particular stability of (CuCl)3. From the figure, there is one attractive spike (−0.023 a.u.) and two repulsive spikes (0.008 and 0.024 a.u.) in (CuCl)4 cluster. Obviously, the interactions of (CuCl)3 cluster are much stronger than those of (CuCl)4 cluster, indicating (CuCl)3 is more stable than (CuCl)4 cluster, which is in agreement with the stability predicted by binding energy analysis. Moreover, there is a signature of the attractive attraction between the Cu atoms in (CuCl)3 and (CuCl)4 clusters, which could be inferred from the range of the Cu⋯Cu contacts in them. The distance of Cu⋯Cu contacts in them are 2.65 and 2.76 Å, respectively, and both of which are smaller than the sum of their van der Waals radii (ca. 2.80 Å).63 It is evident that the distance of Cu⋯Cu contacts in (CuCl)3 cluster is smaller than that of (CuCl)4, which gives extra support to the NCI analysis.
The Ag/Cl interactions in (AgCl)6 cluster is characterized by a blue region in the isosurface, indicating an attractive interaction. Importantly, the NCI method reveals that the lengthening of the Cu⋯Cu distance from (CuCl)3 (2.65 Å) to (CuCl)6 (2.72 Å) is accompanied by the weakening of the Cu⋯Cu interactions. This is manifested in the Fig. 4a and c plots as the shift of the characteristic Cu⋯Cu peak towards more positive values, namely, changing from 0.029 to 0.026 a.u. Fig. 4c shows the characteristic feature of three spikes in the densities ranging from −0.024 to −0.027 a.u., demonstrating the occurrence of attractive noncovalent attractions, and three spikes in the densities changing from 0.014 to 0.027 a.u. manifesting three repulsive interactions. Three kinds of noncovalent interactions of the Cu⋯Cu interactions in the same (CuCl)3 unit, and Cu⋯Cu and Cu⋯Cl interactions between the two (CuCl)3 units are the blue regions of the (CuCl)6 isosurface. The red regions in the isosurface of (CuCl)6 manifest the steric and coulombic repulsion. The stability of (CuCl)6 indicates that the attractive dispersion interactions of Cu⋯Cu and Cu⋯Cl contacts outweigh the steric and coulombic repulsion.
Electronic and bonding properties of (CuCl)3 and (AgCl)3 clusters
The most relevant valence molecular orbitals of (CuCl)3 and (AgCl)3 clusters are shown in Fig. 5. Perusal of Fig. 5 reveals that all three-membered copper and silver rings exhibit a composite bonding mode. The bonding of (CuCl)3 and (AgCl)3 clusters is characterized by a common ring-shaped electron density, more commonly seen in organic molecules, which is constructed by highly delocalized σ- and π-type MOs. The delocalized σ and π electron density in the rings could be associated with the cyclic delocalization of electron density, which is a characteristic feature of aromaticity. Interestingly, in the peripheral three-membered rings there are both σ- and π-type MOs (HOMO-15, HOMO-23 for (CuCl)3 cluster, and HOMO-20, HOMO-27 for (AgCl)3 cluster) resulting from the bonding combination of the 3d atomic orbitals (AO) of Cu and 4d AOs of Ag atoms in the rings with the 3p AOs of Cl atoms, respectively. The electronic structure and bonding properties of the three-membered Cu and Ag rings closely resemble those of the aromatic Au rings in Au3Cl3 cluster.64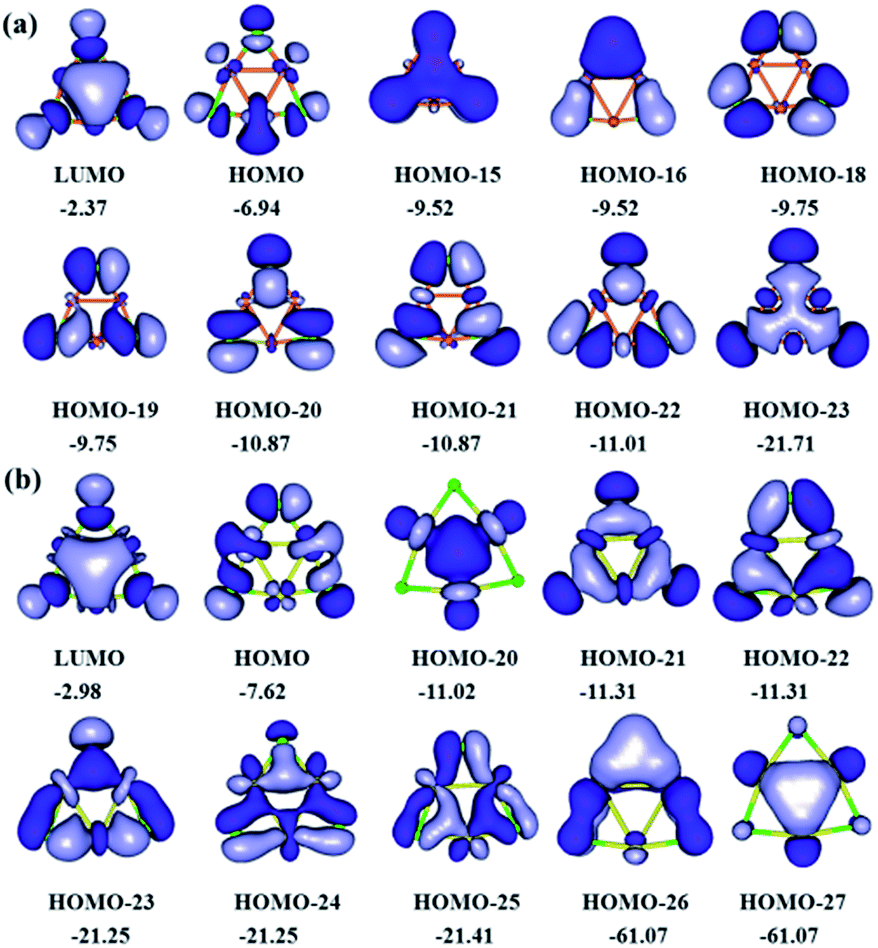 | ||
| Fig. 5 Most relevant valence molecular orbitals of (a) (CuCl)3 and (b) (AgCl)3 clusters (isocontour value of 0.02 a.u.; eigenvalues in eV). | ||
Aromaticity
Planarity, high stability, bond length equalization, and HOMO–LUMO gaps are conventionally good indicators of aromaticity.65 Nucleus-independent chemical shift (NICS) value50 is the most widely used as a quantitative measure for aromaticity. Negative NICS values indicate aromaticity, while positive values imply antiaromaticity. NICS values are the negative of absolute magnetic shielding tensors. In 2001, Boldyrev et al. reported the experimental and theoretical evidence of aromaticity in Al42−, an all-metal compound.66 Tsipis et al. have investigated the aromatic properties in gold and silver rings of MnHn (M = Ag, Au; n = 3–6) molecules and the ligand-stabilized aromatic three-membered gold rings in a series of cyclo-Au3LnH3−n (L = CH3, NH2, OH and Cl; n = 1–3).64,65 The DFT study of “all-metal” aromatic compounds has been reviewed by Tsipis.67 All-metal aromaticity and antiaromaticity, and the aromaticity in transition-metal systems have been reported.68,69 Stanger introduced the NICS-scan method,70 which is analogous to the aromatic ring center shieldings approach. Here, we employ NICS-scan method to analyze the aromaticity of the tube clusters. Considering that the out-of-plane component of NICS-scan, NICSzz-scan, is a better index of aromaticity,71,72 the aromatic properties along z-axes (coined as NICSzz-scan) within the range of −4.0–4.0 Å above the geometric centers of the clusters are calculated. NICS(0) positions of NICSzz-scan curves are the geometric centers of the clusters. Fig. 6 plots the NICSzz-scan curves for (AgCl)3, (CuCl)3, (CuCl)6 and (CuCl)9 clusters. It can be seen that all the molecules exhibit negative NICS(0) values, and the NICS values obtained at various positions inside, above or outside the molecular frame proved the existence of diatropic ring-current effect, characteristic for aromaticity. Based on the figure, (AgCl)3 cluster is generally less aromatic than (CuCl)n clusters. Noteworthy is that the NICS(0) value of (AgCl)3 (−11.3 ppm) is smaller than that of (CuCl)3 (−14.6 ppm), and both of which are smaller than the that of Au3Cl3 cluster (−23.8 ppm).64 From the figure, we can see that the curves oscillate. Concretely, there is one trough of wave in the NICSzz-scan curves (red and black curves) of (AgCl)3 and (CuCl)3 clusters, respectively. There are two and three troughs of wave in the NICSzz-scan curves (blue and green curves) of (CuCl)6 and (CuCl)9 clusters, respectively. Analysis of the data obtained, it is obviously that the aromaticity of the center of the (MCl)3 unit is higher than the neighboring positions along z-axis of the cluster. It is concluded that the curves oscillate due to the (MCl)3 unit. In Fig. 6, the (MCl)3 units displayed in dotted oval, and the aromaticity brought by the (MCl)3 unit can be clearly seen. The NICS-scan curves of (CuCl)3, (CuCl)6 and (CuCl)9 clusters exhibit one, two and three dotted ovals, respectively. In a word, the (MCl)3 unit can be proved existing based on NICS-scan method.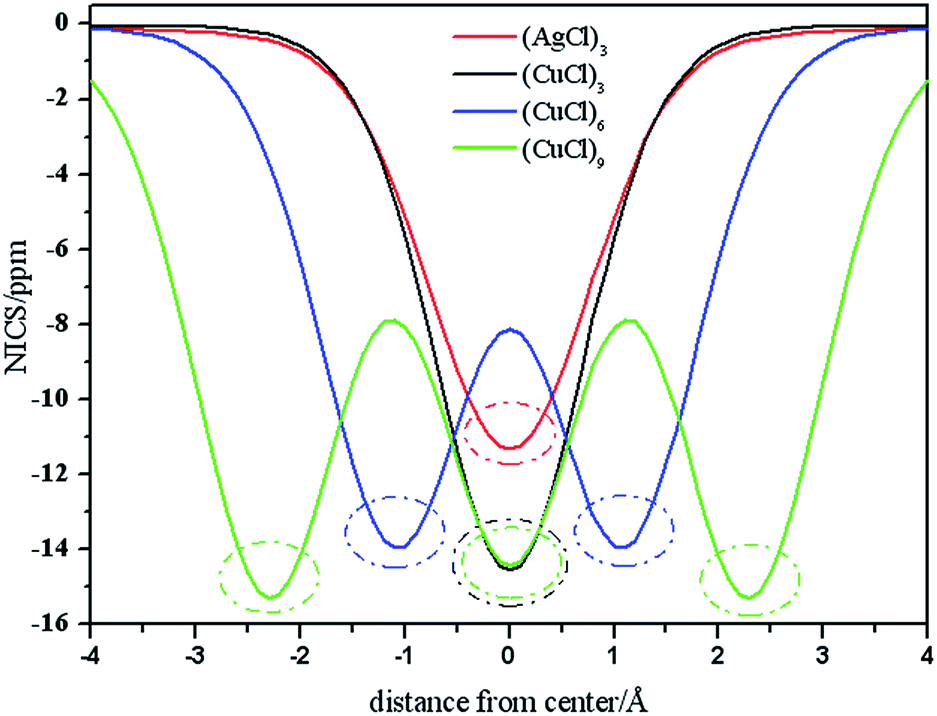 | ||
| Fig. 6 The NICSzz-scan curves for (AgCl)3, (CuCl)3, (CuCl)6, (CuCl)9 and (CuCl)12 within the range of −4.0 to 4.0 Å above the geometric centers of the clusters. | ||
Conclusions
In present work, the geometries of (MCl)n (n = 1–12, M = Cu and Ag) clusters are investigated using the method combining GA with TPSSh functional. The relevant low-lying structures are obtained. The computational results indicate that the structures of (CuCl)n and (AgCl)n clusters share many similarities. The global minima structures are planar and nonplanar cyclic structures in the range of n = 1–5. At n = 7, a distorted low symmetric structure is the most stable isomer. At n = 8, the windmill structure is the global minimum. When n = 3, 6, 9 and 12, (AgCl)n clusters are more stable than neighboring clusters, whereas (CuCl)n clusters are more stable than neighboring clusters at n = 3, 6, 8, 10 and 12. The four three-member-ring structure intersecting cubic structure is the most stable isomer for (CuCl)10, whereas the catenane isomer is most stable for (AgCl)10 cluster. The trimer is very stable with high binding energy. A new folding manner, tube conformation, is found for (CuCl)n and (AgCl)n clusters. When n is the integral multiple of three and the structure includes triangular units, it exhibits aromatic. The NICS-scan method and NCI index are used to character the clusters.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21273008, 21573001), the Natural Science Foundation of Anhui Provincial University (2015KJ013) and the financial support of the Fuyang Teachers College (2015FSKJ12). The calculations are carried out on the High-Performance Computing Centre of Anhui University.References
- J. M. L'Hermite, F. Rabilloud, P. Labastie and F. Spiegelman, Eur. Phys. J. D, 2001, 16, 77–80 CrossRef.
- F. Rabilloud, O. Bonhomme, J. M. L'Hermite and P. Labastie, Chem. Phys. Lett., 2008, 454, 153–157 CrossRef CAS.
- P. Koirala, M. Willis, B. Kiran, A. K. Kandalam and P. Jena, J. Phys. Chem. C, 2010, 114, 16018–16024 CAS.
- F. Rabilloud and D. Mathian, J. Cluster Sci., 2012, 23, 165–176 CrossRef CAS.
- Q. Wang, Q. Sun and P. Jena, J. Chem. Phys., 2009, 131, 124301 CrossRef CAS PubMed.
- F. Rabilloud, J. Comput. Chem., 2012, 33, 2083–2091 CrossRef CAS PubMed.
- N. Shao, Y. Pei, Y. Gao and X. C. Zeng, J. Phys. Chem. A, 2009, 113, 629–632 CrossRef CAS PubMed.
- H. Grönbeck, M. Walter and H. Häkkinen, J. Am. Chem. Soc., 2006, 128, 10268–10275 CrossRef PubMed.
- L. Cheng, Y. Yuan, X. Zhang and J. Yang, Angew. Chem., Int. Ed., 2013, 52, 9035–9039 CrossRef CAS PubMed.
- Y. Yuan, L. Cheng and J. Yang, J. Phys. Chem. C, 2013, 117, 13276–13282 CAS.
- J. M. López de Luzuriaga, M. Monge, M. E. Olmos and D. Pascual, Organometallics, 2015, 34, 3029–3038 CrossRef.
- G. R. Desiraju, J. Chem. Soc., Dalton Trans., 2000, 3745–3751 RSC.
- A. Taubert, Angew. Chem., 2004, 116, 5494–5496 CrossRef.
- M. C. Li, H. Yu, R. Huang, F. Bai, M. Trevor, D. D. Song, B. Jiang and Y. F. Li, Nanoscale Res. Lett., 2013, 8, 1–6 CrossRef PubMed.
- H. Daupor and S. Wongnawa, Mater. Chem. Phys., 2015, 159, 71–82 CrossRef CAS.
- Y. H. Yin, H. S. Chen and Y. Song, J. Mol. Struct.: THEOCHEM, 2010, 959, 30–34 CrossRef CAS.
- H. C. Müller Rösing, A. Schulz and M. Hargittai, J. Am. Chem. Soc., 2005, 127, 8133–8145 CrossRef PubMed.
- F. Rabilloud, J. Phys. Chem. A, 2012, 116, 3474–3480 CrossRef CAS PubMed.
- F. Rabilloud, F. Spiegelman, J. L'Hermite and P. Labastie, J. Chem. Phys., 2001, 114, 289–305 CrossRef CAS.
- A. Kovács and R. J. Konings, J. Mol. Struct., 2002, 643, 155–160 CrossRef.
- H. Zhang and Z. Schelly, J. Phys. Chem. A, 2000, 104, 6287–6294 CrossRef CAS.
- S. Glaus and G. Calzaferri, Photochem. Photobiol. Sci., 2003, 2, 398–401 CAS.
- P. Schwerdtfeger, R. P. Krawczyk, A. Hammerl and R. Brown, Inorg. Chem., 2004, 43, 6707–6716 CrossRef CAS PubMed.
- A. Walsh, C. R. A. Catlow, R. Galvelis, D. O. Scanlon, F. Schiffmann, A. A. Sokol and S. M. Woodley, Chem. Sci., 2012, 3, 2565–2569 RSC.
- D. M. Deaven and K. M. Ho, Phys. Rev. Lett., 1995, 75, 288–291 CrossRef CAS PubMed.
- R. L. Johnston, Dalton Trans., 2003, 4193–4207 RSC.
- A. Shayeghi, D. Götz, J. Davis, R. Schaefer and R. L. Johnston, Phys. Chem. Chem. Phys., 2015, 17, 2104–2112 RSC.
- B. Pinter, L. Broeckaert, J. Turek, A. Růžička and F. De Proft, Chem.–Eur. J., 2014, 20, 734–744 CrossRef CAS PubMed.
- A. Chakraborty, K. K. Ramachandran, S. S. Yamijala, S. K. Pati and T. K. Maji, RSC Adv., 2014, 4, 35167–35170 RSC.
- M. Hapka, M. Dranka, K. Orłowska, G. Chałasiński, M. M. Szczęśniak and J. Zachara, Dalton Trans., 2015, 44, 13641–13650 RSC.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori Sanchez, J. Contreras Garcia, A. J. Cohen and W. T. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- H. L. Hermann, G. Boche and P. Schwerdtfeger, Chem.–Eur. J., 2001, 7, 5333–5342 CrossRef CAS.
- E. Wächtler, S. H. Privér, J. R. Wagler, T. Heine, L. Zhechkov, M. A. Bennett and S. K. Bhargava, Inorg. Chem., 2015, 54, 6947–6957 CrossRef PubMed.
- L. Ren, L. J. Cheng, Y. Feng and X. M. Wang, J. Chem. Phys., 2012, 137, 014309 CrossRef PubMed.
- L. J. Cheng and J. L. Yang, J. Chem. Phys., 2013, 138, 141101 CrossRef PubMed.
- L. F. Li and L. J. Cheng, J. Chem. Phys., 2013, 138, 094312 CrossRef PubMed.
- Y. Yuan and L. Cheng, J. Chem. Phys., 2012, 137, 044308 CrossRef PubMed.
- Z. M. Tian and L. J. Cheng, Phys. Chem. Chem. Phys., 2015, 17, 13421–13428 RSC.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- S. Hamad, C. Catlow, S. Woodley, S. Lago and J. Mejias, J. Phys. Chem. B, 2005, 109, 15741–15748 CrossRef CAS PubMed.
- M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. P. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision B. 01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 CrossRef CAS.
- J. Sugar and A. Musgrove, J. Phys. Chem. Ref. Data, 1990, 19, 527–616 CrossRef CAS.
- H. P. Loock, L. M. Beaty and B. Simard, Phys. Rev. A, 1999, 59, 873 CrossRef CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. V. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- K. P. Huber and G. Herzberg, Constants of diatomic molecules, Springer, 1979 Search PubMed.
- F. H. de Leluw and A. Dymanus, J. Mol. Spectrosc., 1973, 48, 427–445 CrossRef.
- T. Martin and H. Schaber, J. Chem. Phys., 1980, 73, 3541–3546 CrossRef CAS.
- K. A. Kacprzak, O. Lopez-Acevedo, H. Häkkinen and H. Gronbeck, J. Phys. Chem. C, 2010, 114, 13571–13576 CAS.
- M. R. Wiseman, P. A. Marsh, P. T. Bishop, B. J. Brisdon and M. F. Mahon, J. Am. Chem. Soc., 2000, 122, 12598–12599 CrossRef CAS.
- Y. Liu, Z. M. Tian and L. J. Cheng, RSC Adv., 2016, 6, 4705–4712 RSC.
- C. Priest, Q. Tang and D. E. Jiang, J. Phys. Chem. A, 2015, 119, 8892–8897 CrossRef CAS PubMed.
- S. Darby, T. V. Mortimer-Jones, R. L. Johnston and C. Roberts, J. Chem. Phys., 2002, 116, 1536–1550 CrossRef CAS.
- J. Contreras García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. T. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- L. López, P. Ruiz, M. Castro, J. Quijano, M. Duque-Noreña, P. Pérez and E. Chamorro, RSC Adv., 2015, 5, 62946–62956 RSC.
- J. Soto-Delgado, J. Torras, L. J. del Valle, F. Estrany and C. Alemán, RSC Adv., 2015, 5, 9189–9203 RSC.
- Y. Q. Feng and L. J. Cheng, RSC Adv., 2015, 5, 62543–62550 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- A. C. Tsipis and C. A. Tsipis, J. Am. Chem. Soc., 2005, 127, 10623–10638 CrossRef CAS PubMed.
- C. A. Tsipis, E. E. Karagiannis, P. F. Kladou and A. C. Tsipis, J. Am. Chem. Soc., 2004, 126, 12916–12929 CrossRef CAS PubMed.
- X. Li, A. E. Kuznetsov, H. F. Zhang, A. I. Boldyrev and L. S. Wang, Science, 2001, 291, 859 CrossRef CAS PubMed.
- C. A. Tsipis, Coord. Chem. Rev., 2005, 249, 2740–2762 CrossRef CAS.
- A. I. Boldyrev and L. S. Wang, Chem. Rev., 2005, 105, 3716 CrossRef CAS PubMed.
- T. R. Galeev and A. I. Boldyrev, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2011, 107, 124–147 RSC.
- A. Stanger, J. Org. Chem., 2006, 71, 883–893 CrossRef CAS PubMed.
- P. V. R. Schleyer, M. Manoharan, H. Jiao and F. Stahl, Org. Lett., 2001, 3, 3643–3646 CrossRef CAS.
- C. Corminboeuf, T. Heine, G. Seifert, P. von Ragué Schleyer and J. Weber, Phys. Chem. Chem. Phys., 2004, 6, 273–276 RSC.
| This journal is © The Royal Society of Chemistry 2016 |